





De Novo Sequencing of Proteins by Mass Spectrometry: Workflow, Confidence, and Practical Limits
- a novel peptide not represented in sequence databases
- undocumented substitutions or engineering changes
- species-poor or incomplete reference databases
- unexpected processing or truncation
- a post-translational modification (PTM) pattern that disrupts routine matching
- Are the b ions or y ions continuous across the region that matters?
- Are the mass gaps uniquely assignable to residues?
- Does the precursor ion charge state support informative fragmentation?
- Is there visible interference, co-isolation, or a likely chimeric spectrum?
- Do replicate spectra support the same residue path?
- a full confident peptide sequence
- one or more high-confidence sequence tags
- a partial peptide with defined residue ambiguity
- peptide-level evidence for a variant, truncation, or modification pattern
- a preliminary protein-level reconstruction model
De novo sequencing of proteins by mass spectrometry is most useful when the question is narrower than “determine the entire protein sequence.” In the right setting, LC-MS/MS can produce strong residue-level evidence, sequence tags, or peptide-level calls without depending on a correct reference database. In harder projects, the output is usually a ranked sequence hypothesis with stated gaps, ambiguity, and a follow-up confirmation plan, not a complete primary-structure assignment.
In de novo sequencing, residue order is inferred directly from tandem mass spectrometry fragment ion patterns instead of starting with a database-search candidate. That makes the approach useful for novel peptides, undocumented engineering changes, impurity bands, and modified analytes that do not match expected sequence space very well. It also changes how confidence should be judged. The key question is not just whether a peptide-spectrum match exists, but whether fragment ion continuity, sequence tag length, residue ambiguity, PTM localization logic, and orthogonal validation all point in the same direction.
Quick Decision Guide
Use de novo sequencing first when the sample is relatively clean, the precursor ion can be isolated well, and the project needs peptide-level sequence evidence or a discriminating sequence tag.
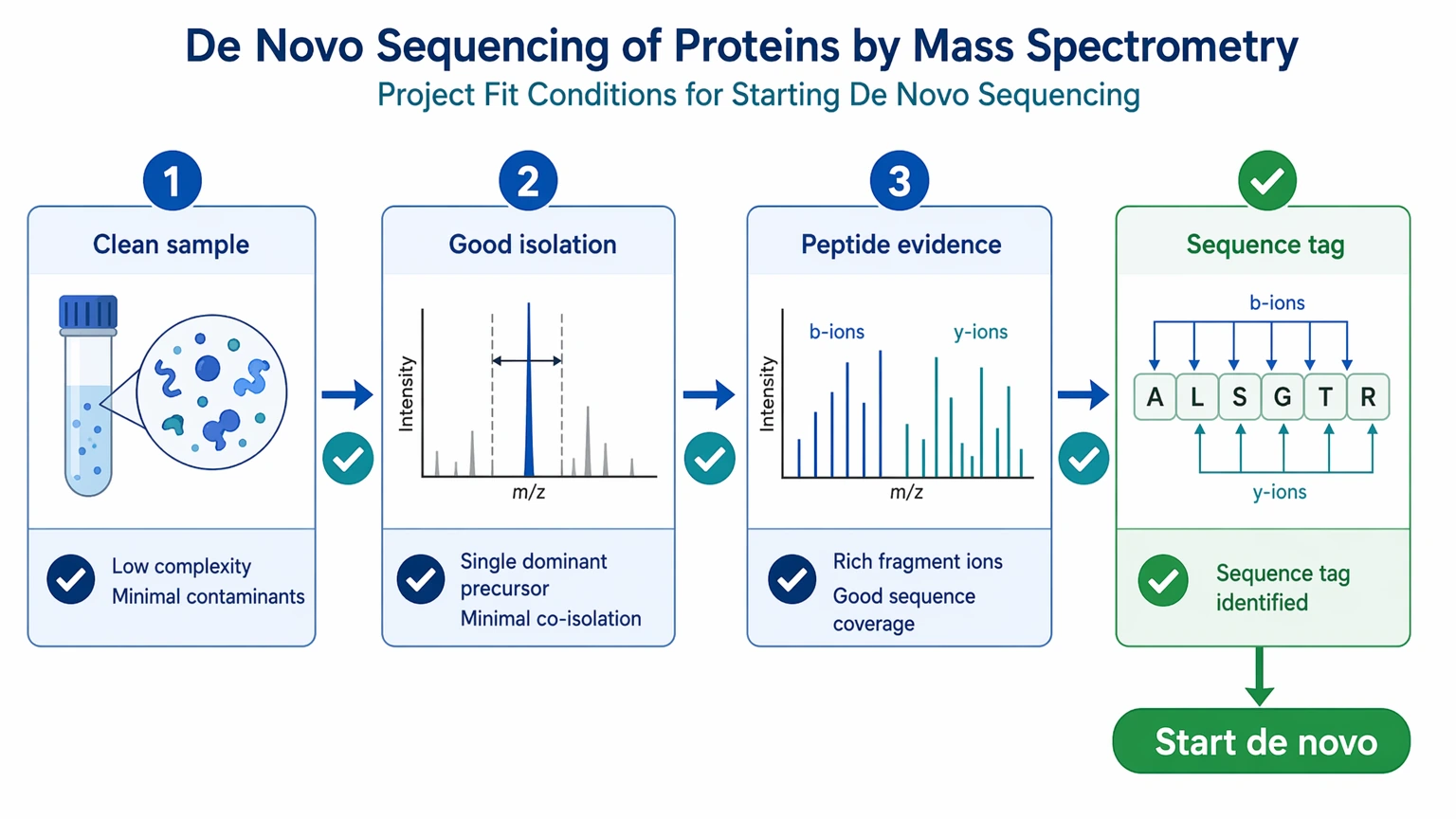
Use it cautiously when the sample is mixed, low in amount, highly modified, or expected to require protein-level reconstruction from incomplete peptide evidence.
Plan follow-up confirmation from the start if the decision depends on terminal completeness, leucine/isoleucine resolution, exact PTM localization, or full-length protein reconstruction.
What De Novo Sequencing Actually Answers
De novo sequencing is often described as if it directly reads a whole protein. Most LC-MS/MS workflows do not work that way. In bottom-up proteomics, the starting point is usually a peptide generated by protease digestion or present as an isolated analyte. A precursor ion is selected, fragmented, and interpreted through mass differences between fragment ions, especially b ions and y ions.
That peptide-level inference can be very informative, but protein-level reconstruction is a separate problem. It depends on multiple overlapping peptides, useful sequence coverage, and a coherent way to assemble the evidence. For many projects, that distinction decides whether the method fits the actual question.
A contaminant band from a gel, a venom peptide fraction, an engineered linker region, or a clipped therapeutic fragment may only need one or two strong sequence tags. By contrast, proving the exact full-length sequence of a mixed or PTM-rich protein often requires more than de novo sequencing alone.
When Database Search Is Not Enough
A database search is still efficient when the expected sequence is known and the search space is relevant. De novo sequencing becomes more informative when that assumption breaks down. Typical triggers include:
In these settings, a conventional peptide-spectrum match may be absent, weak, or misleading. De novo sequencing does not remove uncertainty, but it can show where the evidence is truly local and residue-based.
Practical LC-MS/MS Workflow for De Novo Sequencing
The workflow should start with the real decision question. Is the project trying to recover a full peptide sequence, identify a short discriminating sequence tag, test whether a construct differs from expectation, or assemble a protein-level reconstruction? That target changes the sample form, acquisition strategy, and validation burden.
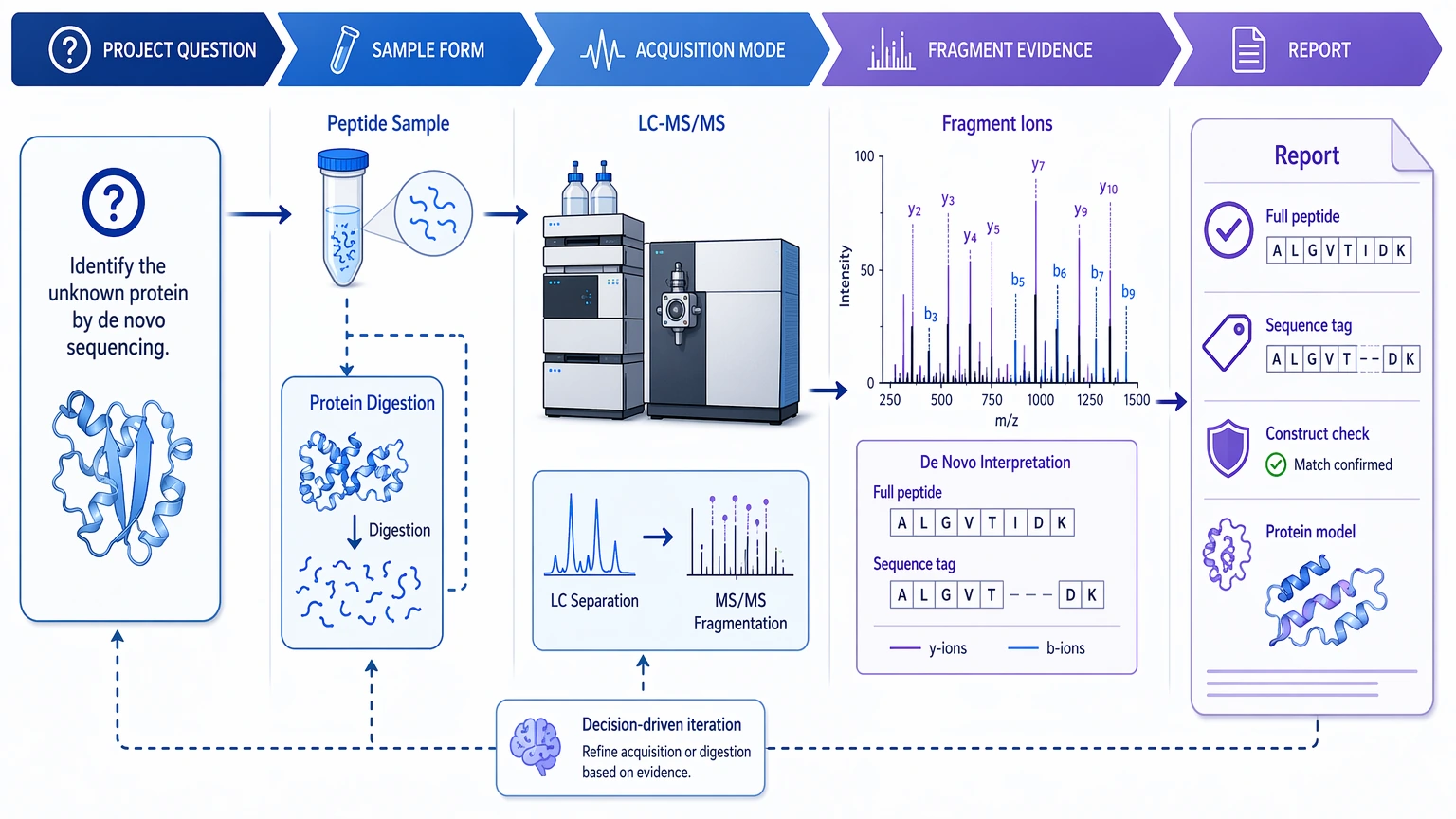
For isolated peptides, direct LC-MS/MS analysis may be appropriate. For intact proteins, the work often moves into bottom-up proteomics through protease digestion, because peptide-scale tandem mass spectrometry is usually easier to interpret than intact-protein fragment maps. Sample purity still matters here. Mixed components raise co-isolation risk and increase the chance of a chimeric spectrum, where fragment ions from more than one precursor ion appear together.
Fragmentation strategy also affects interpretability. Collision-induced dissociation (CID) and higher-energy collisional dissociation (HCD) often produce useful b ions and y ions. Electron-transfer dissociation (ETD) can help with higher-charge peptides or labile PTMs. In some projects, combining HCD or CID with ETD improves local confidence because one fragmentation mode can cover gaps left by another.
The raw output is still not a sequence. It is a fragment-ion pattern that may support several candidate residue paths, one dominant path, or no credible path at all.
The table below helps set expectations before method selection.
| Scenario | Recommended workflow | Key limitation | Validation need |
|---|---|---|---|
| Purified peptide with clean MS/MS | Direct de novo peptide sequencing | Local residue ambiguity may remain | Replicate LC-MS/MS or intact mass |
| Purified protein with unknown sequence | Protease digestion plus peptide-level inference | Protein-level reconstruction may be partial | Multiple proteases and intact mass |
| PTM-rich peptide | HCD with ETD review when possible | PTM localization may interrupt ladder interpretation | Targeted confirmatory MS |
| Gel band or impurity fraction | Cleanup plus LC-MS/MS | Co-migrating proteins may mix evidence | Orthogonal purity check |
| Complex mixture | Pre-fractionation before interpretation | Chimeric spectrum risk is high | Additional separation strategy |
Use this table as a triage tool. It separates cases that are likely to yield a clear peptide call from those more likely to return partial sequence evidence.
Service Routes to Consider
For this project scenario, readers usually compare these service routes before requesting a quote or submitting samples.
How Confidence Is Built from Fragment Evidence
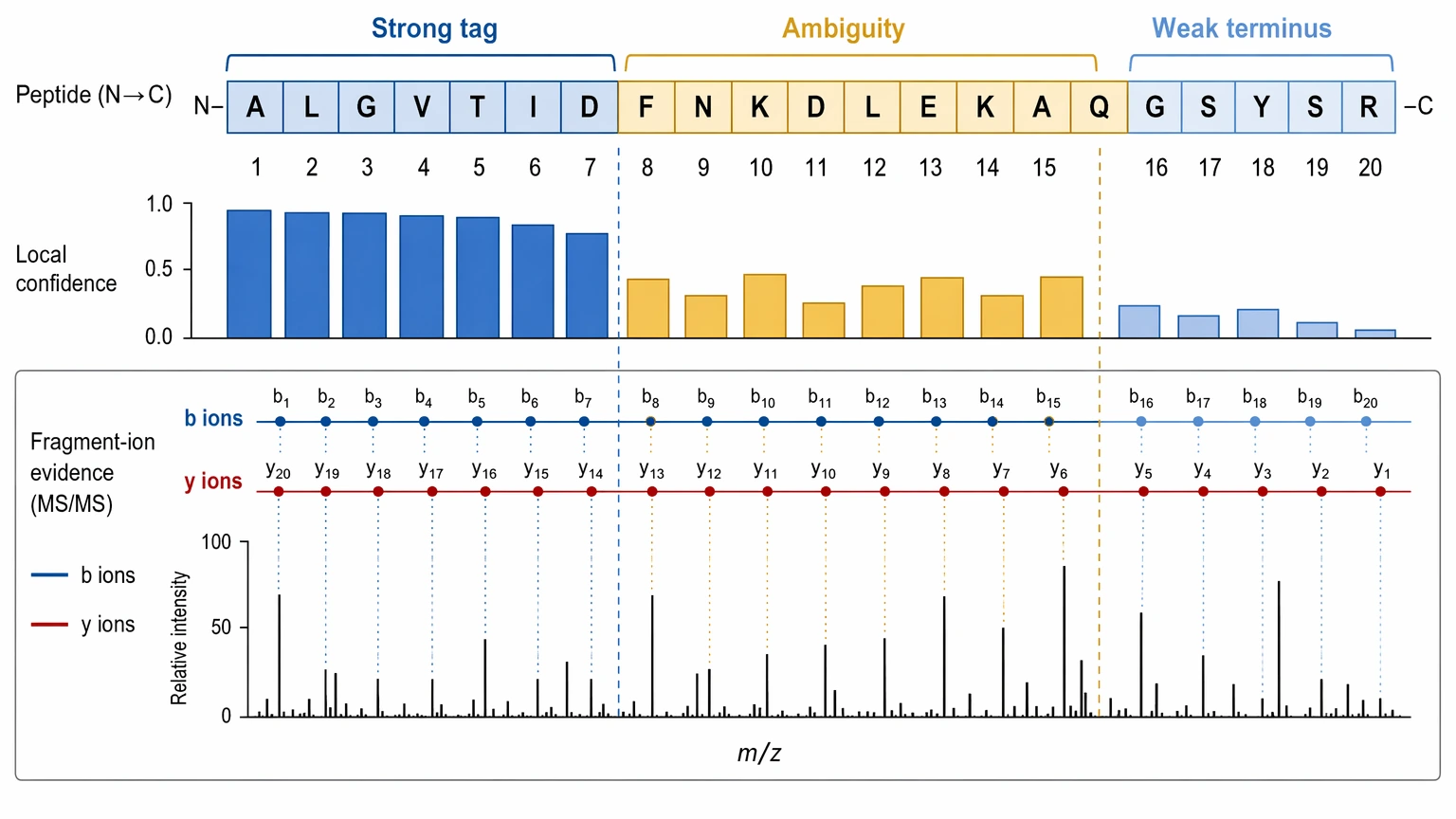
Confidence in de novo sequencing is local before it becomes global. A proposed sequence may contain one well-supported tag, one stretch with residue ambiguity, and one weakly supported terminus. Treating the whole sequence as equally certain hides how the evidence is actually distributed.
The most useful questions are practical:
A strong report should describe local confidence scoring, not just one sequence-level score. It should also separate a confident sequence tag from a full confident peptide call.
One important limitation is that standard LC-MS/MS often cannot resolve leucine/isoleucine ambiguity directly because those residues are isobaric. Another is that PTMs can either help interpretation through a diagnostic mass shift or make it harder by interrupting fragment-ion continuity. When PTMs, incomplete ladders, or database-search limits are involved, one boundary should stay explicit: MS/MS-based de novo sequencing can leave some residue positions, PTM localization sites, or terminal regions unresolved even when the overall sequence hypothesis is plausible.
Peptide-Level Inference Versus Protein-Level Reconstruction
Readers often ask whether peptide-level de novo sequencing can be treated as direct protein sequencing. Usually, it should not.
Peptide-level inference asks whether a specific precursor ion supports a candidate residue order. Protein-level reconstruction asks whether many peptide observations can be assembled into a defensible full or partial protein model. The second task depends on peptide overlap, digestion coverage, missing regions, termini, PTMs, and whether the sample contains one protein species or several related forms.
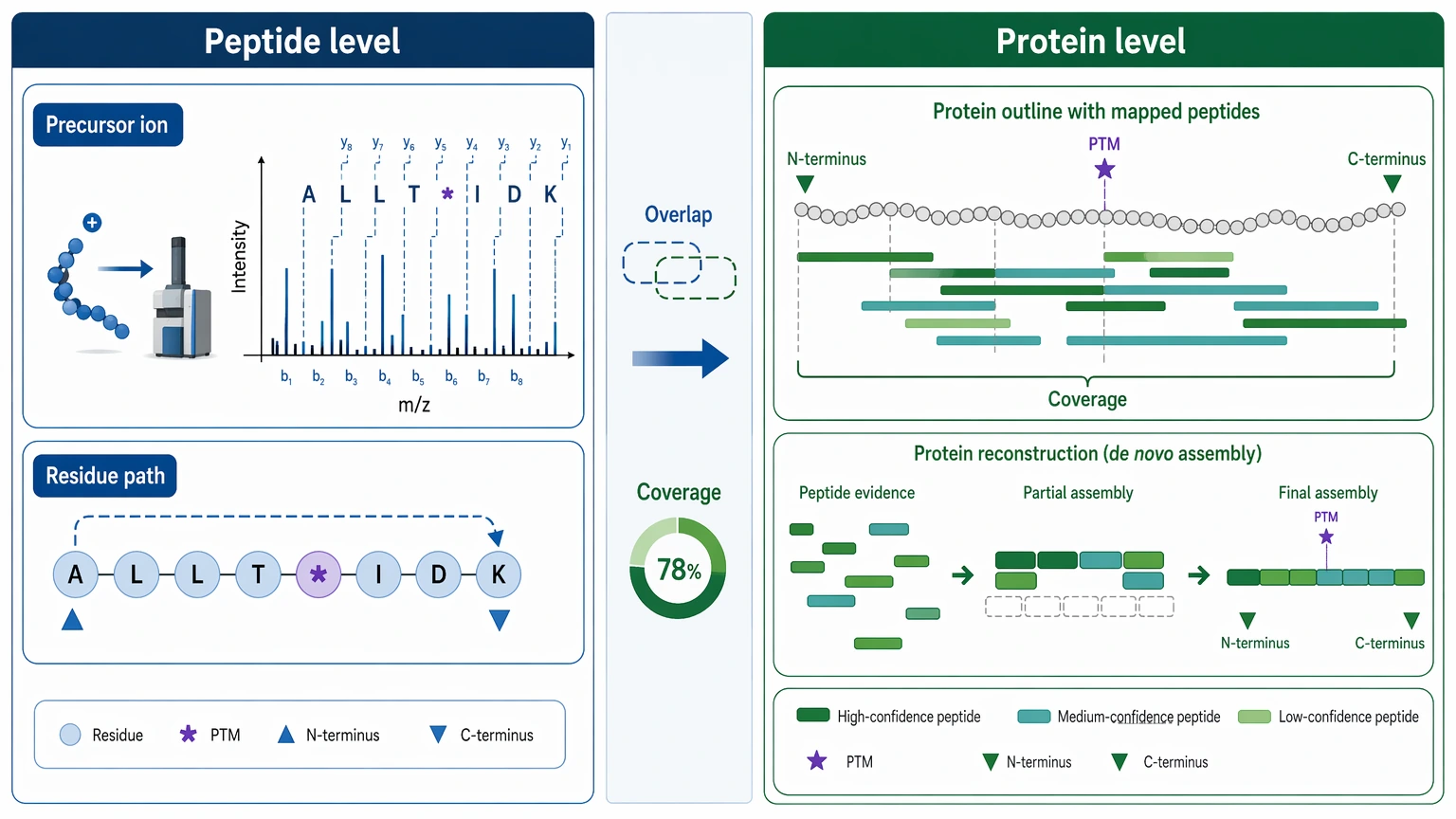
That is why full protein claims need careful limits. A set of strong de novo peptides may show that a protein family, engineered region, truncation site, or impurity source is identifiable. The same dataset may still fall short of proving exact termini, isoform status, or complete residue-by-residue primary structure.
Expected Results and Validation Methods
A realistic deliverable is not always a complete sequence. Common immediate deliverables include:
Immediate deliverables should be separated from follow-up confirmation. A good project plan states what can be reported directly from LC-MS/MS evidence and what still needs orthogonal validation.
| Evidence class | Immediate deliverable | Follow-up confirmation |
|---|---|---|
| Continuous b/y ion ladder | Local residue order in that segment | Replicate acquisition or alternate fragmentation |
| Long sequence tag | Discriminating peptide or family-level clue | Database-constrained review or overlapping peptides |
| Intact mass agreement | Global consistency with a candidate model | Peptide-level confirmation of disputed regions |
| Alternative protease digestion | Overlapping support across sequence regions | Assembly review for protein-level reconstruction |
| Targeted confirmatory MS | Testing of specific disputed positions or PTM sites | Integration into the final interpretation |
If your project needs a feasibility read before you commit sample, you can submit your requirements to MtoZ Biolabs to evaluate the workflow, sample state, and expected report format against the characterization question you actually need to answer.
Key Cautions and Practical Limits
Several limits matter more than a long list of generic workflow risks.
Sample quality or amount limits. Low-abundance peptides may produce weak fragment ion series. Impure fractions may yield co-isolated precursors, especially in gel bands, bioactive fractions, or stressed product samples.
Controls and repeat expectations. A single spectrum is rarely enough when a residue position is disputed. Replicate LC-MS/MS, alternative protease digestion, or targeted confirmatory runs are often needed when the conclusion will affect downstream decisions.
Batch or contamination risk. Mixed protein backgrounds, carryover, and chimeric spectra can create sequence paths that look chemically plausible but actually merge evidence from more than one precursor ion.
Interpretation boundaries. Sequence coverage does not equal full certainty. Bottom-up de novo sequencing may support one region strongly while leaving termini, residue ambiguity, or PTM localization partly open.
When another method is the better next step. If the project depends on exact terminal definition, intact proteoform resolution, or confirmation of a short unresolved region, top-down proteomics, intact mass analysis, targeted site validation, or Edman-style terminal logic may be more informative than collecting more of the same bottom-up data.
Conclusion
De novo sequencing of proteins by mass spectrometry works best as a question-fit workflow for sequence inference, not as an automatic whole-protein readout. It is most convincing when clean precursor selection, informative tandem mass spectrometry, coherent fragment-ion ladders, and orthogonal validation all support the same interpretation. It becomes less decisive when the sample is mixed, the fragmentation pattern is sparse, PTMs interrupt continuity, or protein-level reconstruction is expected from limited peptide evidence. For unknown peptides, engineered sequence changes, impurity bands, or PTM-rich characterization projects, the most useful next step is to define the exact decision target and the minimum acceptable confidence boundary. If you want to compare workflow options or validation paths for a real sample, contact MtoZ Biolabs to discuss sample type, amount, and report goals before you commit to de novo sequencing.
FAQ
Can de novo sequencing identify a protein if only one peptide is interpretable?
Sometimes, but only under narrow conditions. One strong sequence tag may be enough to assign a protein family, detect a construct mismatch, or flag an impurity source. It is usually not enough to support full protein-level reconstruction.
Does a high de novo score mean the sequence is correct at every residue?
Not necessarily. Confidence can vary across a peptide. A strong central tag may sit next to uncertain termini or unresolved leucine/isoleucine ambiguity, so the score should be read together with fragment-ion coverage.
Are multiple proteases useful even when the main goal is de novo sequencing?
Yes. Different digestion patterns can create overlapping peptides that improve sequence coverage and help test whether a proposed protein-level reconstruction is internally consistent.
Can de novo sequencing distinguish an amino acid substitution from a PTM?
Sometimes, but not automatically. A mass shift may fit either explanation until fragment placement, PTM localization, and orthogonal evidence narrow the possibilities.
What is the most common reason a promising sample still yields weak sequence evidence?
Poor precursor purity is a frequent problem. Even when signal intensity looks acceptable, co-isolation can generate a chimeric spectrum that weakens confidence in the residue path.
What should a decision-ready de novo sequencing report include?
Look for precursor ion details, fragment-ion evidence, sequence tags, residue ambiguity, PTM localization status, sequence coverage, confidence scoring by region, and a clear statement of which findings are direct deliverables versus follow-up confirmation.
How to order?

