





De Novo Sequencing Method vs Database Search: Which Strategy Fits Novel or PTM-Rich Peptides?
- Sample composition: Is the material relatively clean, or are co-eluting components likely?
- Reference quality: Does the organism, construct, or peptide design have dependable sequence coverage?
- Modification expectations: Are PTMs known and limited, or open-ended and structurally diverse?
- Deliverable type: Do you need a candidate list, a sequence tag, a prioritized structure proposal, or confirmation-ready evidence?
- Follow-up capacity: Can the project support orthogonal validation such as targeted LC-MS/MS, intact mass confirmation, or synthetic peptide comparison?
Choose database search first when the peptide sequence is likely to exist in a dependable reference and the main job is to confirm known peptides or a narrow range of expected variants. Choose the de novo sequencing method first when the sample may contain a novel peptide, a noncanonical peptide, undocumented processing products, or modification patterns that make the reference incomplete or misleading. In many LC-MS/MS projects, the most defensible route is a hybrid workflow that pairs fast peptide-spectrum match (PSM) assignment with targeted direct sequence interpretation for spectra that stay unresolved.
A practical screening rule is straightforward: start with database search if reference database completeness is strong and the expected post-translational modification (PTM) set is limited; start with the de novo sequencing method if true sequence novelty is likely or the searchable reference is weak; use a hybrid workflow if you expect both known content and unexplained modified or novel peptide signals. MS/MS evidence can support a sequence proposal, but it does not always resolve leucine/isoleucine ambiguity, every PTM site, or every isomeric interpretation with full certainty.
Why This Comparison Matters for Novel or PTM-Rich Peptides
This decision usually appears after a team has pilot LC-MS/MS data or a defined sample set, but before full interpretation starts. Common situations include peptides from poorly annotated organisms, engineered analogs, degradation fragments, impurity profiling, and signaling peptides with several concurrent modifications. At that stage, the project is no longer asking for a general description of identification methods. It is asking which route is less likely to misclassify the real analyte.
A poor method choice costs more than extra analysis time. A reference-driven workflow can return plausible hits even when the real peptide is absent from the database. A de novo-first plan can produce long candidate lists that look promising even though the entries do not all carry the same evidential weight. The key issue is which method fits the novelty risk, the PTM burden, and the quality of the MS/MS data actually in hand.
Head-to-Head Comparison
The table below summarizes the main tradeoffs before the deeper review.
| Dimension | Database Search | De Novo Sequencing Method |
|---|---|---|
| Reference requirement | Requires adequate sequence representation | Does not require an exact database entry |
| Best fit | Known targets, annotated organisms, limited expected variation | Novel peptide discovery, noncanonical peptide content, weak references |
| PTM handling | Effective for expected PTMs, but variable modification space can expand quickly | Can expose unexplained mass patterns, but PTM interpretation may remain partial |
| Confidence model | PSM score with false discovery rate control | Sequence-level confidence score, sequence tag continuity, fragment support |
| Main failure mode | Correct peptide missing from the search space | Sparse or mixed fragment ions, interrupted ion ladders, unresolved isobaric residues |
| Typical output | Identified peptides with modification candidates | Sequence proposals, sequence tags, prioritized candidate backbones |
| Validation burden | Often moderate for known peptides | Usually higher for novel or PTM-rich assignments |
So the choice should come back to four practical questions: Is the correct backbone present in the search space? How quickly does the PTM search space grow? Can the MS/MS spectrum support residue-level interpretation? What level of evidence does the project need at the end?
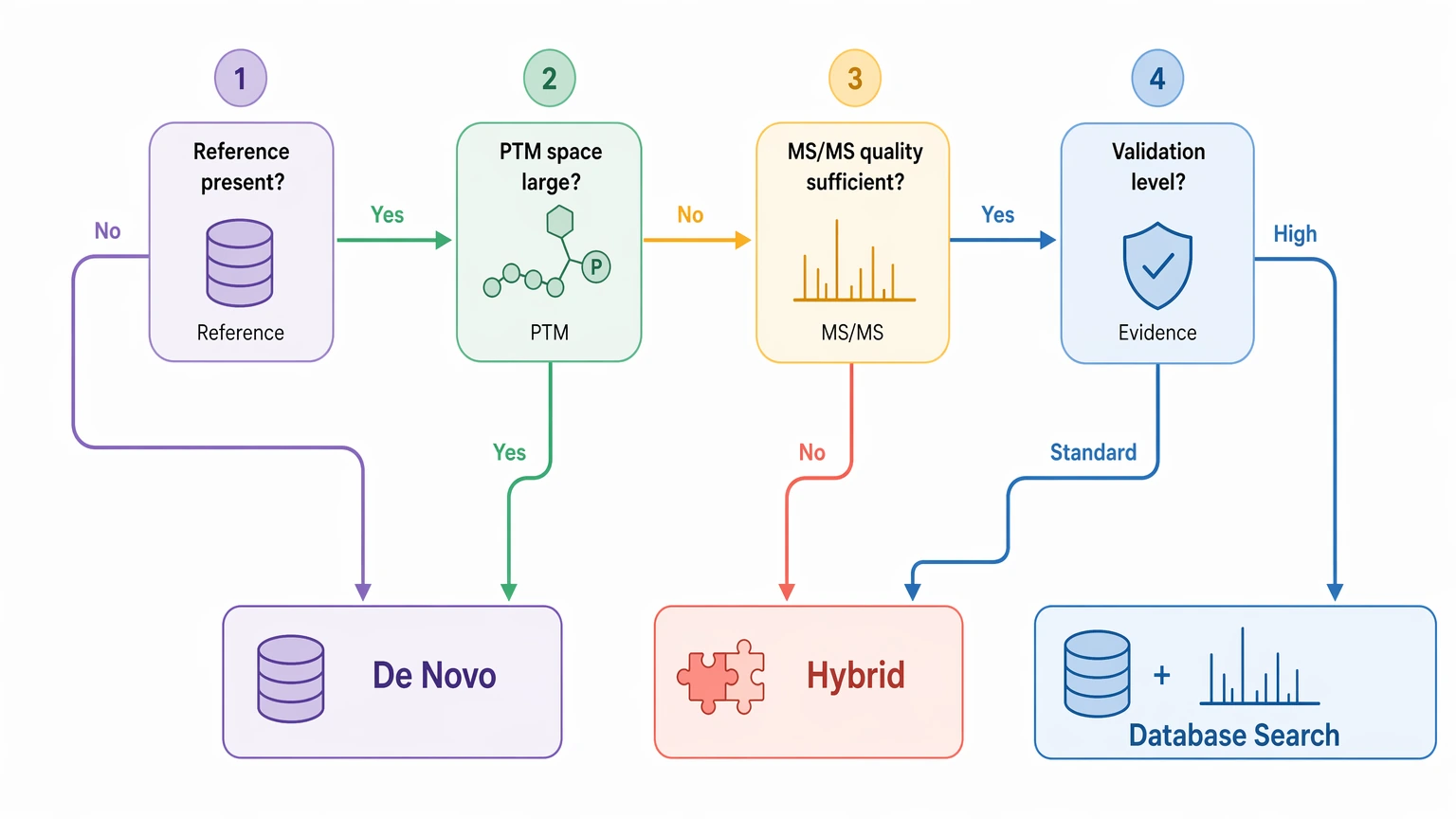
The Four Factors That Usually Decide the Workflow
Reference dependence and novelty risk
Database search works best when the correct sequence is already represented, or when a small set of expected substitutions and variable modification settings can approximate it. In a well-annotated proteome, that is efficient and often sufficient.
That logic breaks down when the analyte is outside the searchable universe. A strain-specific sequence, undocumented cleavage product, synthetic analog, or unexpected impurity may simply not exist in the database. If that happens, the algorithm can only return the closest available explanation. A statistically acceptable PSM is not the same thing as a biologically correct answer.
The de novo sequencing method addresses that problem by building sequence evidence directly from fragment ions instead of relying on prior sequence inclusion. That makes it more suitable when the project is truly asking what is present rather than which known peptide fits best.
PTM burden and search-space expansion
PTM-rich peptides can push standard search settings beyond their comfortable range. A focused search can handle expected modifications such as oxidation or phosphorylation. The difficulty rises when the sample may contain several PTMs per peptide, labile PTMs, or unexpected combinations that rapidly expand the search space.
Each added variable modification increases the number of candidate explanations for a spectrum. That can weaken false discovery rate control, create competing site assignments, and make PTM localization less stable. An open search can help flag unexplained mass shifts, but it does not by itself tell you whether the mass difference reflects a modification, a substitution, or both.
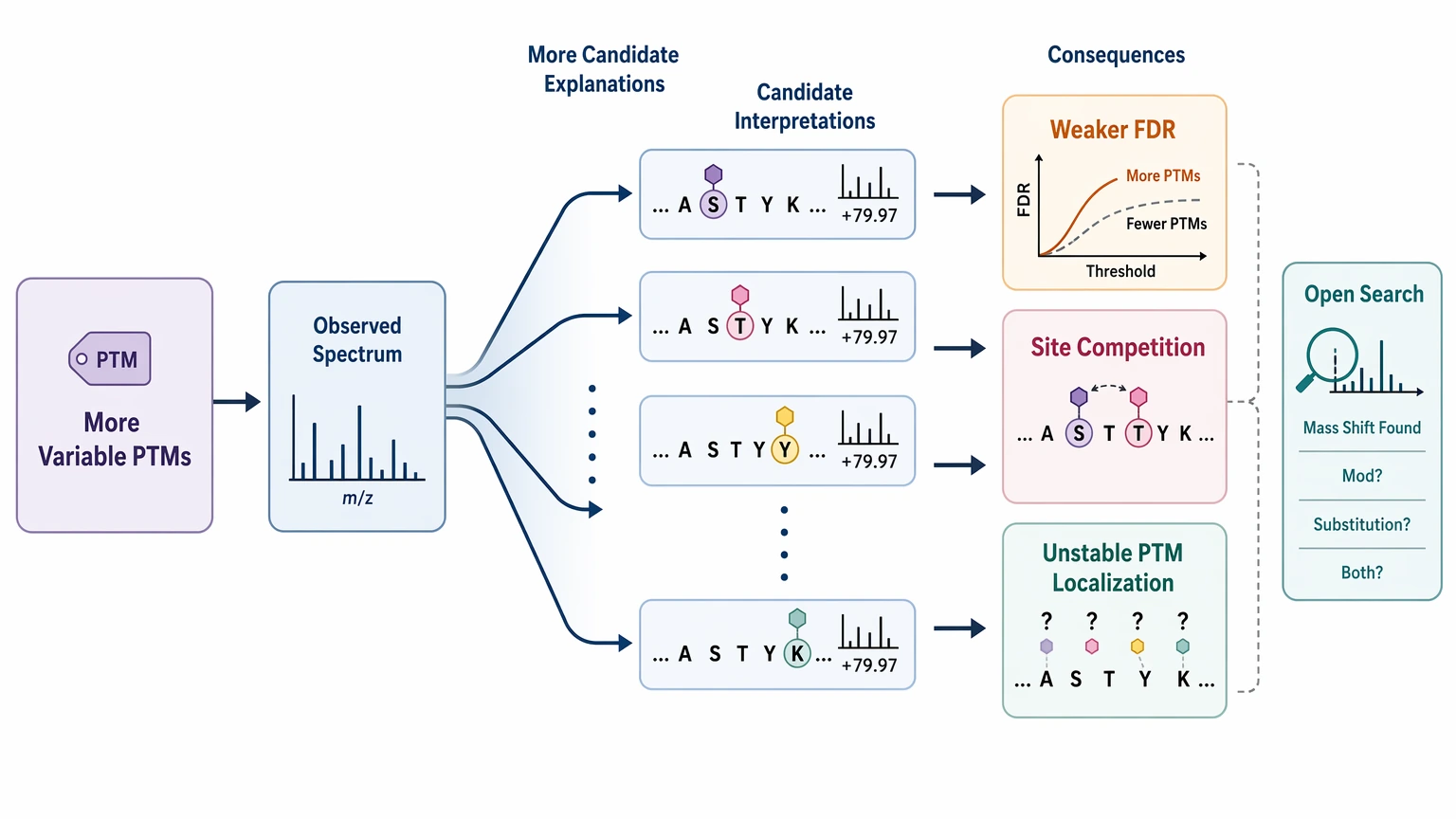
The de novo sequencing method can help show where a reference-based model stops matching the data, especially when a sequence tag is supported but the full reference assignment is not. A practical limit remains: MS/MS alone may not localize every PTM site with confidence, particularly for labile modifications or spectra with incomplete ion ladders.
MS/MS interpretability
The de novo sequencing method is only as strong as the underlying tandem mass spectrometry evidence. Before making it the primary strategy, check whether the spectra truly support residue-by-residue inference instead of broad pattern matching.
The table below highlights the signal features that matter most when deciding whether direct sequence interpretation is realistic.
| MS/MS feature | Why it matters | Effect on strategy choice |
|---|---|---|
| Accurate precursor mass | Constrains candidate sequence explanations | Supports both workflows |
| Strong fragment mass accuracy | Narrows residue and modification interpretation | Supports both workflows |
| Continuous b-ion series or y-ion series | Improves sequence path continuity | Strongly favors de novo sequencing method |
| Low co-isolation interference | Reduces mixed-spectrum ambiguity | Strongly improves confidence |
| Replicate spectral support | Helps separate recurrent signals from one-off noise | Strengthens validation planning |
If the dataset shows broken ladders, dominant neutral losses, or frequent co-isolation, de novo analysis may still add value, but the realistic output may be a partial sequence or a high-value sequence tag rather than a fully confirmed peptide structure.
Deliverable confidence and validation burden
Method choice should match the kind of decision the project needs to support. A discovery-stage program may accept ranked candidates. A development-stage program may need assignments that are ready for confirmation.
A database search result is usually presented as a PSM with score and false discovery rate support. A de novo result needs a more layered readout: local confidence score, sequence coverage, backbone continuity, PTM evidence strength, and any remaining isobaric residues. For live sequencing or PTM-interpretation decisions, you can submit your requirements to MtoZ Biolabs to review LC-MS/MS quality, reference coverage, and the level of confirmation needed before choosing a de novo or validation workflow.
Service Routes to Consider
When Each Strategy Fits Best
Choose database search first
Use database search when the sample comes from a well-annotated organism, the peptide family is already known, and the expected PTM set is narrow. This route is efficient when the main goal is confirmation rather than discovery. Its main risk is missing true novelty that falls outside the search space.
Choose the de novo sequencing method first
Use the de novo sequencing method when backbone uncertainty is the main analytical problem. That includes poorly annotated species, engineered peptides, unknown degradation products, and impurity or side-product studies where the correct sequence may not exist in the database. Its main risk is overreading sparse spectra as full-length sequences.
Choose a hybrid workflow
For many real projects, a hybrid design is the most balanced option. Start with database search to assign spectra that fit known biology. Then route unresolved or weakly matched spectra into de novo sequencing method, open search, or sequence tag-guided review. This staged approach is especially useful when a sample may contain both expected peptides and genuinely novel or heavily modified species.
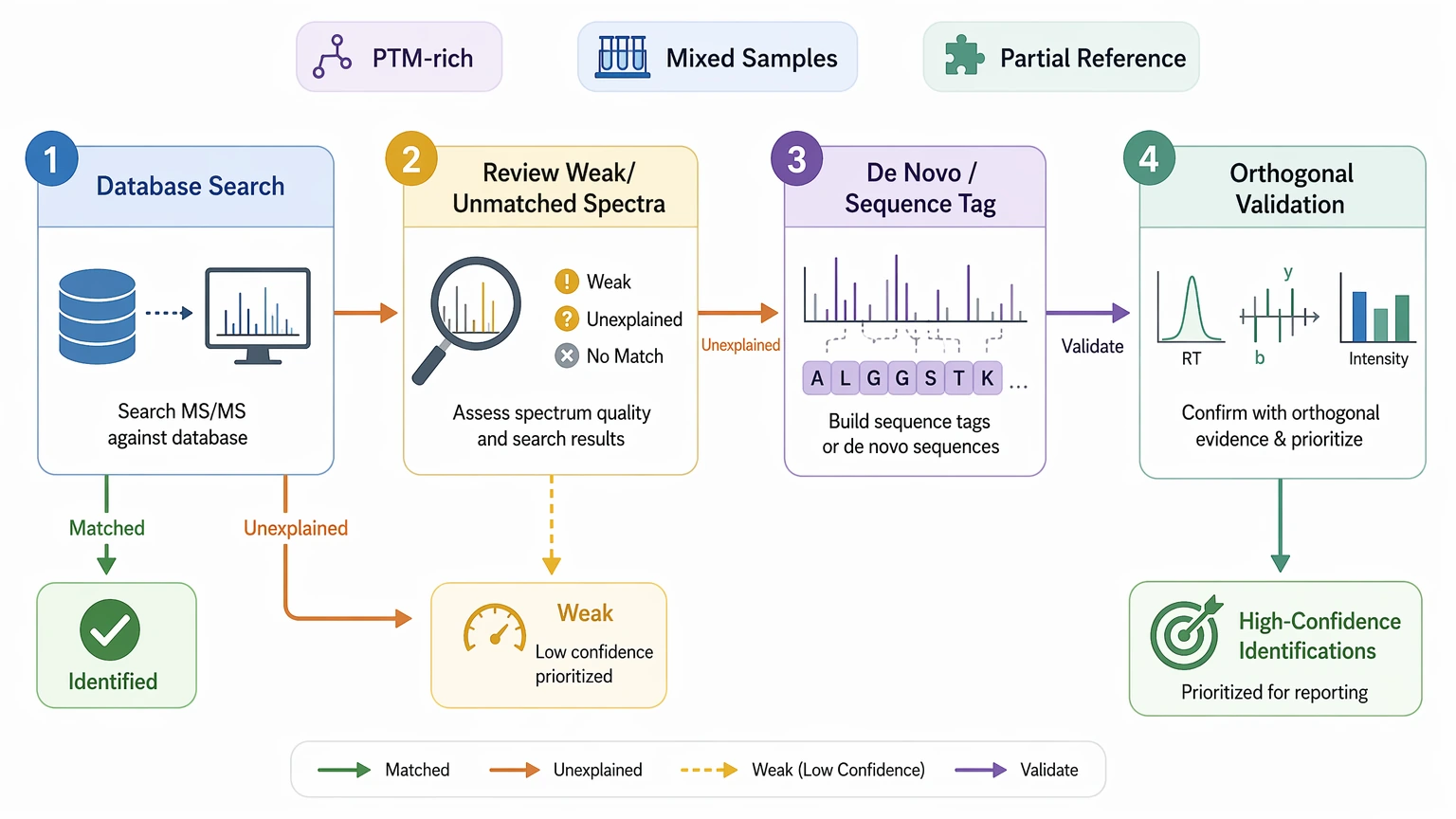
Data and Sample Checks Before You Commit
Before locking in a workflow, define the project in practical terms: sample composition, reference quality, modification expectations, deliverable type, and follow-up capacity. Novelty alone does not determine method fit. A highly novel peptide with weak MS/MS evidence may still call for a staged plan rather than a pure de novo-first approach.
How to Read the Final Report Realistically
A strong report should separate what the data support directly from what still needs confirmation. For difficult unknowns, the most useful deliverable may be one or more candidate sequences, local confidence score maps, PTM hypotheses with explicit localization limits, evidence-backed sequence tags, and a short validation plan for the most decision-relevant targets. That style is more defensible than presenting a single overconfident answer for spectra that remain ambiguous.
Technical Summary and Next Step Guidance
Database search remains the right first move when the reference is trustworthy and the peptide question is narrow. The de novo sequencing method becomes more appropriate when the sample may contain a novel peptide, a noncanonical peptide, or PTM complexity that pushes reference-based interpretation beyond a reliable range. A hybrid workflow is often the most practical choice when known and unknown components are likely to coexist in the same LC-MS/MS dataset.
For synthetic analog programs, degradation studies, modified peptide characterization, or discovery projects with incomplete references, a useful project brief includes sample type, expected sequence background, LC-MS/MS status, suspected PTMs, and the confidence threshold needed for the downstream decision. If you need to evaluate your project or contact us about sequence assignment and validation planning, MtoZ Biolabs can review that workflow context and map it to database search, de novo sequencing method, or a staged confirmation path.
FAQ
Can a hybrid workflow begin with de novo sequencing instead of database search?
Yes. That is reasonable when early evidence suggests the correct peptide is absent from the reference. De novo interpretation can generate sequence tags first, and those tags can later guide restricted database review or targeted confirmation.
What usually makes PTM localization less certain in MS/MS data?
Labile PTMs, missing bracketing ions around the modified residue, mixed precursor isolation, and several plausible site placements can all leave localization supported but not definitive.
Are longer peptides always better candidates for de novo sequencing?
No. Longer peptides may provide more fragment information, but they can also fragment unevenly. A shorter peptide with a clean b-ion series and y-ion series may be easier to interpret.
When is intact mass confirmation especially helpful?
It is especially helpful when a proposed sequence carries multiple modifications, when terminal processing is uncertain, or when several candidate backbones explain the MS/MS evidence almost equally well.
How should teams prioritize spectra for manual review?
Review spectra first if they are reproducible across runs, have strong precursor definition, show interpretable ion ladders, and sit near the project's key decision point, such as a suspected impurity or unmatched dominant signal.
Can database search still contribute when a peptide is novel?
Yes. Even without identifying the exact peptide, it can rule out known alternatives, reveal partial homology, flag modification patterns, and separate truly unexplained spectra from those that only need narrower search settings.
Related Services
Main Service |
Supporting Service |
Validation Service |
Alternative Service |
How to order?

