





De Novo Peptide Sequencing vs Database Search: How to Choose for Novel, Modified, or Proprietary Peptides
- Choose database search first if you expect a valid peptide-spectrum match (PSM) from a known or shareable sequence space.
- Choose de novo peptide sequencing first if the sequence is unknown, confidential, or altered beyond routine search assumptions.
- Choose both together if the peptide is short, PTM-rich, chemically engineered, or important enough to require orthogonal validation before a project decision.
- Is the peptide represented in a usable search space?
- Can the expected chemistry be modeled without creating an unmanageable search problem?
- Does the LC-MS/MS data support residue-by-residue interpretation?
- What level of confidence is needed for the next project step?
- disclosed synthetic peptides,
- expected variants from a design library,
- controlled truncation series,
- samples with a narrow set of known modifications.
- confidential peptide assets,
- unknown impurities,
- natural peptides from poorly annotated sources,
- samples with sequence changes outside routine search assumptions.
- a high-confidence PSM from database search,
- a proposed sequence with local confidence patterns,
- a sequence tag,
- a ranked candidate list,
- a modification map with qualified uncertainty.
- targeted LC-MS/MS against the proposed sequence,
- comparison with a synthetic standard,
- intact mass agreement checks,
- focused testing of the modification model,
- repeat acquisition under cleaner isolation or alternative fragmentation conditions.
- Sample quality and amount: low abundance, mixed components, or poor cleanup can reduce interpretability before any algorithm comes into play.
- Controls and repeat expectations: important assignments often need repeat spectra, alternate charge states, or targeted confirmation rather than one acquisition alone.
- Batch and contamination risk: co-isolated species, carryover, and formulation background can create misleading fragment ladders.
- Interpretation boundaries: short peptides, isobaric residue ambiguity, and unmodeled chemistry can leave uncertainty at specific positions even when the overall candidate is plausible.
- When another method is the better next step: if the primary question is molecular mass confirmation, targeted lot checking, or comparison against a known standard, a simpler targeted or intact-mass workflow may answer it more directly than open-ended sequencing.
Use database search when the peptide is present in a trustworthy reference database or a focused custom FASTA, and when the expected modifications can be defined in the search space. Use de novo peptide sequencing when the peptide is missing from available databases, structurally unusual, or constrained by confidentiality. Use a hybrid workflow when a novel peptide, a proprietary peptide, or a heavily modified candidate needs stronger support than either method is likely to provide on its own.
Quick Decision Block
The distinction matters because the two methods answer different questions. Database search asks whether an observed MS/MS spectrum matches a candidate peptide from a defined sequence collection. De novo peptide sequencing asks whether the observed fragment ion pattern supports a peptide backbone without relying on a complete database. In practice, the better choice is the one that gives you defensible evidence for the next decision, not the one that sounds more advanced.
What Actually Drives the Choice
In peptide work, this choice usually comes up when standard proteomics assumptions stop fitting the sample. The target may be a synthetic impurity, a cyclic peptide, a stapled peptide, a proprietary therapeutic lead, or an unknown natural product with no complete sequence reference.
Four practical questions usually decide the workflow:
If the answer to the first two questions is yes, database search is often the faster and cleaner option. If the answer is no, de novo peptide sequencing becomes much more important. If the fourth answer is “high,” a hybrid plan is often more realistic than expecting one pass to settle the issue.
Side-by-Side Comparison by Project Scenario
The table below works best as a first-pass triage tool.
| Scenario | Best starting workflow | Main constraint | Likely next step |
|---|---|---|---|
| Known peptide in public or internal sequence space | Database search | May miss unexpected truncation or unmodeled PTMs | Confirm only if the result drives a critical decision |
| Proprietary peptide with partial sequence disclosure | Hybrid: restricted custom FASTA plus de novo sequence tag analysis | Hidden sequence regions can weaken PSM confidence | Targeted follow-up or synthetic comparison |
| Unknown natural peptide from incomplete biology | De novo peptide sequencing | Full sequence may not be recoverable from one dataset | Orthogonal validation |
| PTM-rich or chemically modified synthetic peptide | Hybrid | Backbone assignment and PTM localization may diverge | Separate validation of sequence and modification model |
| Mixed or weak spectra | Reacquisition or cleanup before interpretation | Neither method can rescue poor primary evidence | Improve sample or data quality first |
One useful takeaway: for complex peptide samples, “database search vs de novo” is often too simple a frame. The real question is whether the evidence you have supports confirmation, discovery, or a narrower candidate list that still needs follow-up.
When Database Search Is Still the Better Choice
Database search is strongest when the peptide exists in a relevant reference database or custom FASTA, and when the likely PTMs are limited enough to define clearly. In that setting, a strong PSM, sensible false discovery rate (FDR) control, good mass accuracy, and coherent fragment coverage can make peptide identification efficient and interpretable.
This approach is especially practical for:
Its weakness is not just novelty. The deeper issue is search-space dependence. A search engine cannot match what is not there, or what is represented incorrectly. If the peptide contains a noncanonical amino acid, unexpected conjugation, partial cyclization, or an unmodeled mass shift, the top-scoring answer can still be the wrong structural explanation.
Service Routes to Consider
For this project scenario, readers usually compare these service routes before requesting a quote or submitting samples.
When De Novo Peptide Sequencing Becomes Necessary
De novo peptide sequencing is usually the better starting point when the peptide is unknown, absent from the available database, or intentionally withheld from routine sharing. It uses the fragmentation pattern in tandem mass spectrometry data to infer sequence directly from observed mass differences between ions, especially b ions and y ions.
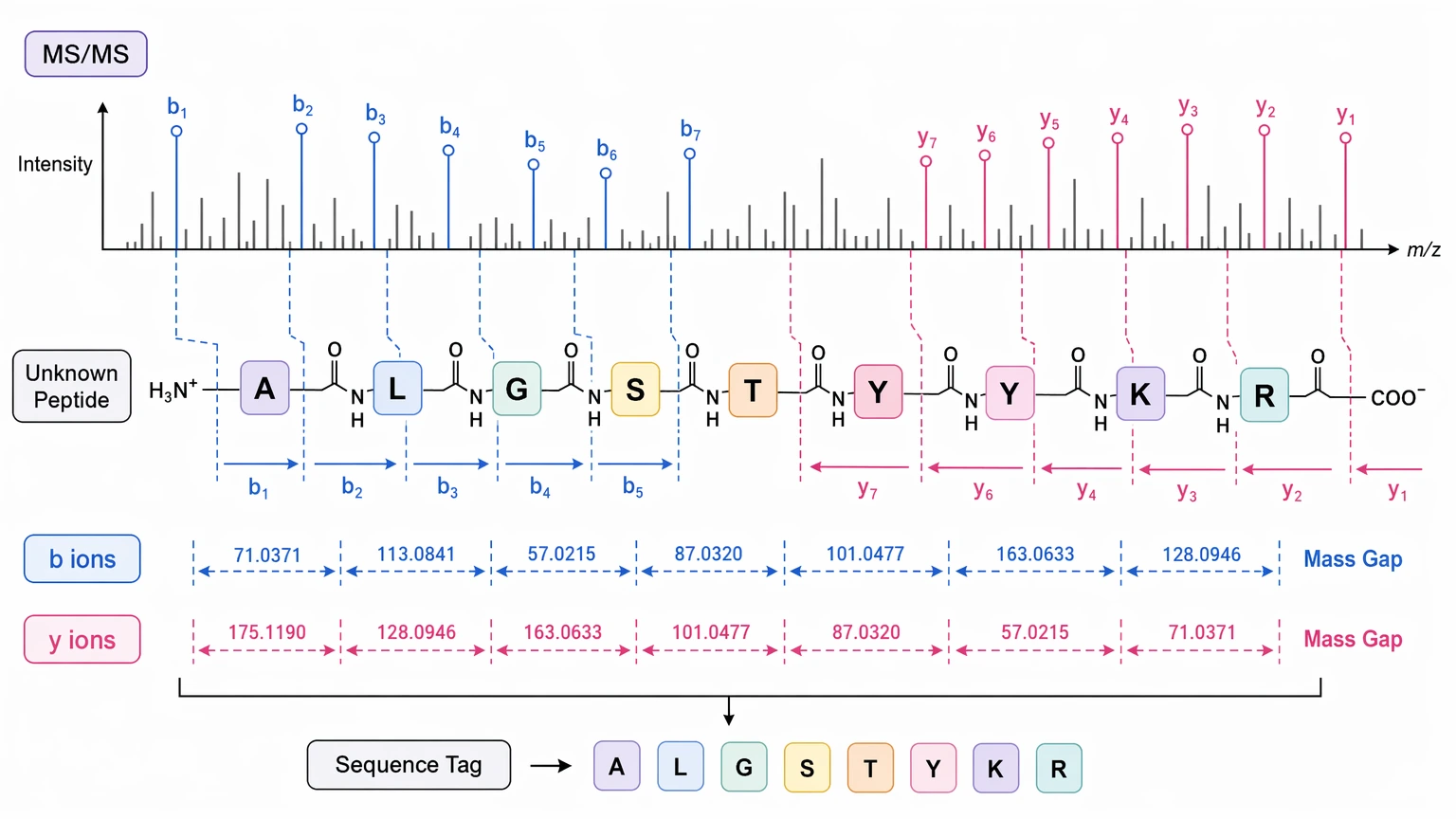
This route often fits:
That said, de novo inference has clear limits. Confidence depends on spectral quality, charge state, precursor purity, ion-series continuity, and whether the peptide fragments in a way that actually supports the backbone. A strong sequence tag can still be useful even when the full sequence remains unsettled. It may narrow synthesis candidates, guide targeted follow-up, or support construction of a restricted custom FASTA.
One limit should be stated directly: tandem MS alone does not always resolve every residue call. Leucine/isoleucine ambiguity is a familiar example, and heavily modified peptides may keep local uncertainty as well. In PTM-rich samples, a de novo result may support only part of the backbone plus a modification mass window, rather than a complete residue-level answer.
Why Modified and Engineered Peptides Complicate Both Methods
Modified peptides stress both workflows, but in different ways. In database search, every extra variable state expands the hypothesis space. A small set of routine events such as oxidation, amidation, or phosphorylation may be manageable. A peptide carrying multiple PTMs, linker chemistry, cyclization, or conjugation is much harder to model cleanly.
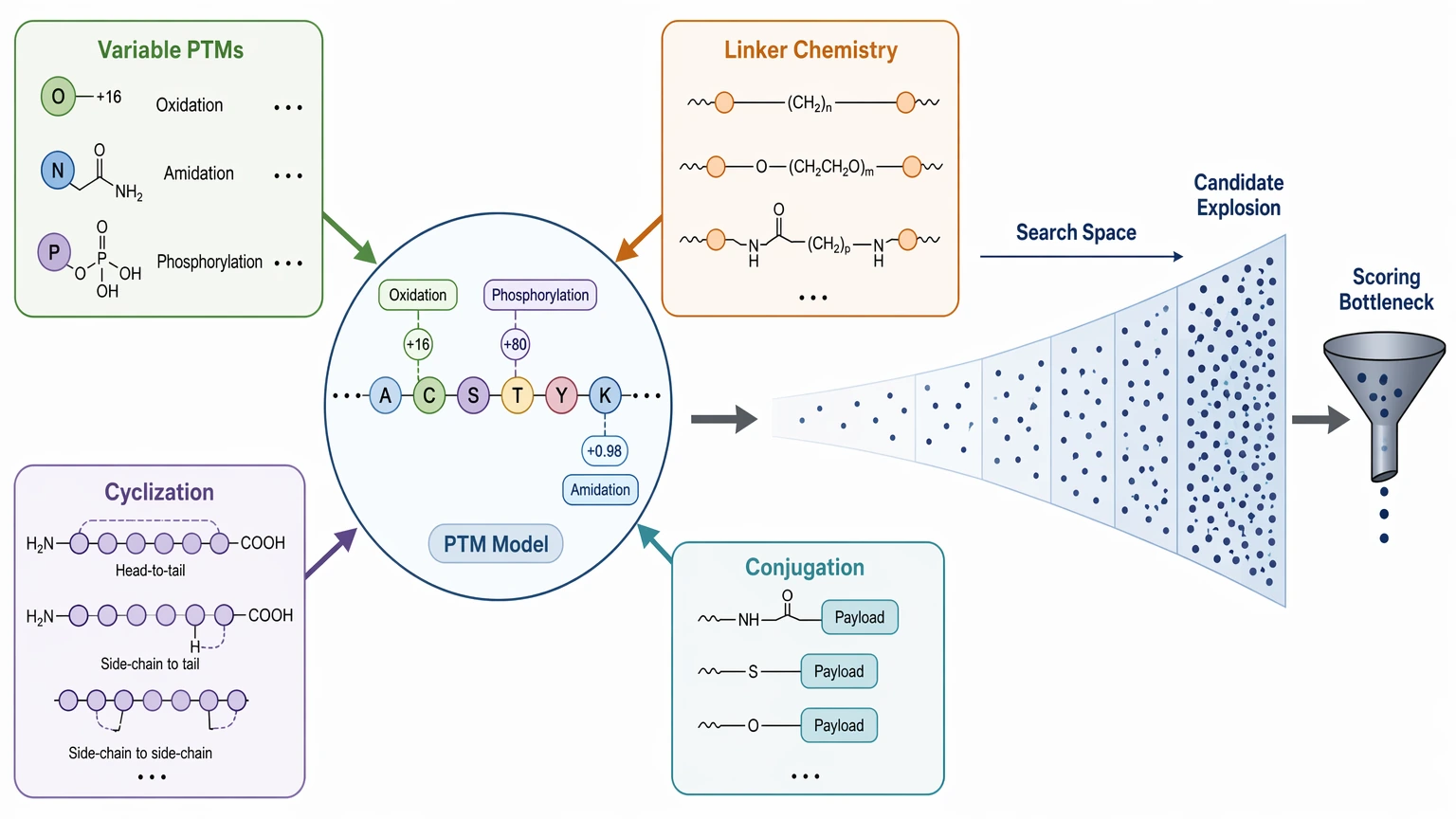
In de novo peptide sequencing, the problem shifts. The backbone may still be partly recoverable, but incomplete fragmentation can blur whether an observed mass shift belongs to one residue, several residues, or a side-chain modification. PTM localization and backbone sequence confidence are related, but they are not the same deliverable.
If your project sits in that gray zone, a practical next step is to submit your requirements to MtoZ Biolabs for review of the sample type, expected chemistry, existing LC-MS/MS files, and whether a de novo-first, database-first, or hybrid interpretation plan is the more defensible choice.
What Evidence Counts as Strong Support
The data should determine how far you trust the answer.
| Evidence type | What it supports | What it does not prove by itself |
|---|---|---|
| Continuous b ions and y ions | Stronger backbone continuity | Exact resolution of every ambiguous residue |
| Clean precursor ion isolation | More reliable fragment attribution | Freedom from all co-isolation artifacts |
| High precursor and fragment mass accuracy | Better residue discrimination | Complete elimination of isobaric ambiguity |
| Reproducible spectra across charge states | Greater confidence in interpretation | Unambiguous PTM localization |
| Intact mass agreement | Overall composition support | Exact sequence order |
The main point is simple: no single metric counts as full proof. Strong peptide identification comes from converging evidence, not one favorable number or one attractive annotation.
Expected Results and Validation Methods
Before choosing a workflow, define the deliverable you actually need.
Immediate deliverables may include:
Follow-up confirmation is a separate stage. It may involve:
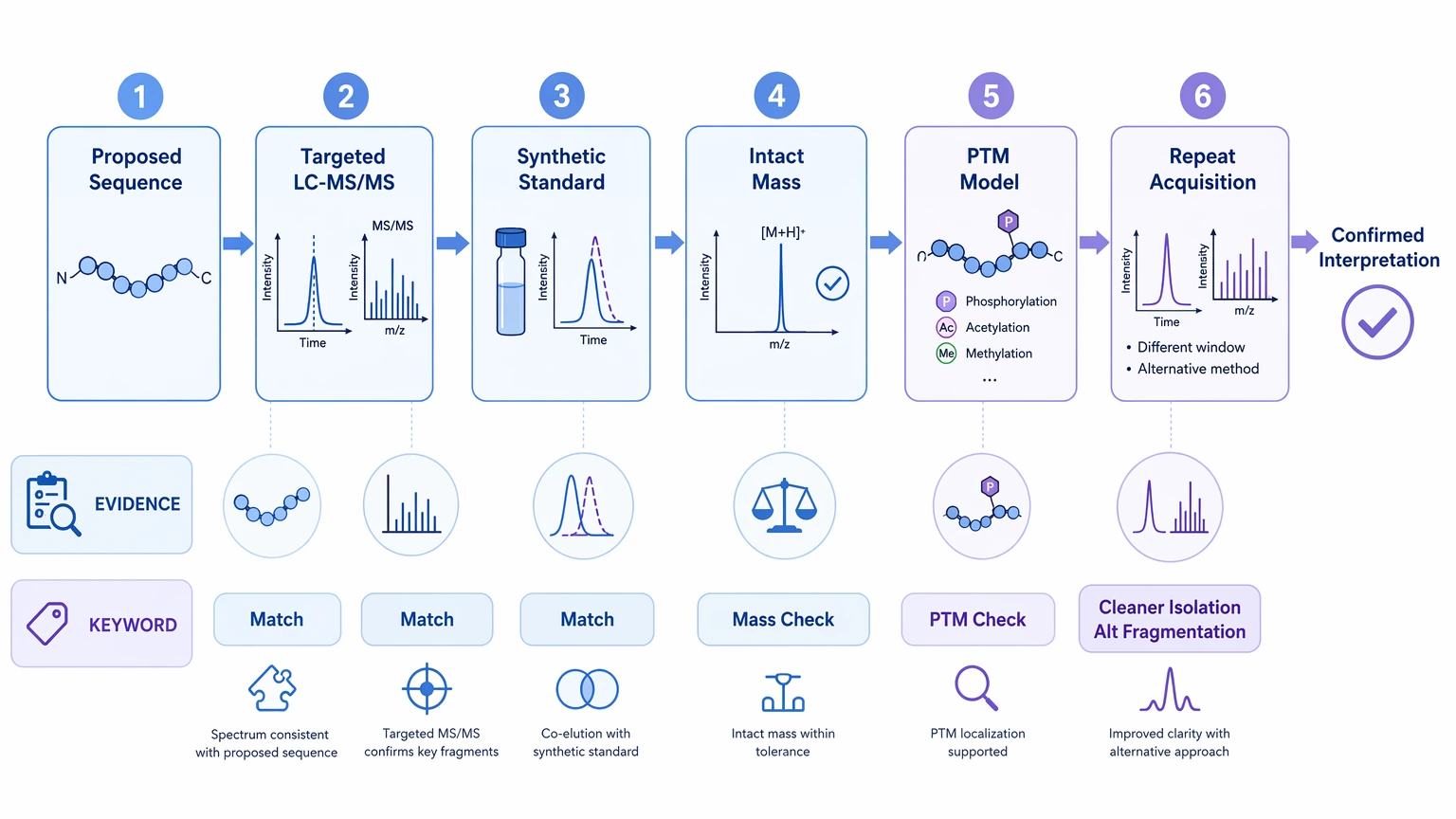
A practical rule helps here: if the result will affect synthesis, intellectual property review, impurity decisions, or downstream assay design, treat the first sequence call as a working interpretation until orthogonal evidence supports it.
A hybrid strategy is often useful at this stage. De novo sequence tags can challenge a weak database assignment, and database search can test whether a de novo proposal is unique within a restricted sequence space.
Key Cautions and Practical Limits
Several limits should be built into planning from the start.
These constraints do not make the methods unreliable. They tell you when reported sequence confidence is strong enough for the decision in front of you, and when the project needs another layer of evidence.
How to Choose for a Live Project
Choose database search when sequence space is available and chemically constrained. Choose de novo peptide sequencing when the peptide is novel, confidential, or structurally outside practical search assumptions. Choose a hybrid workflow when the same project needs both discovery and confirmation logic, especially for modified or proprietary peptides.
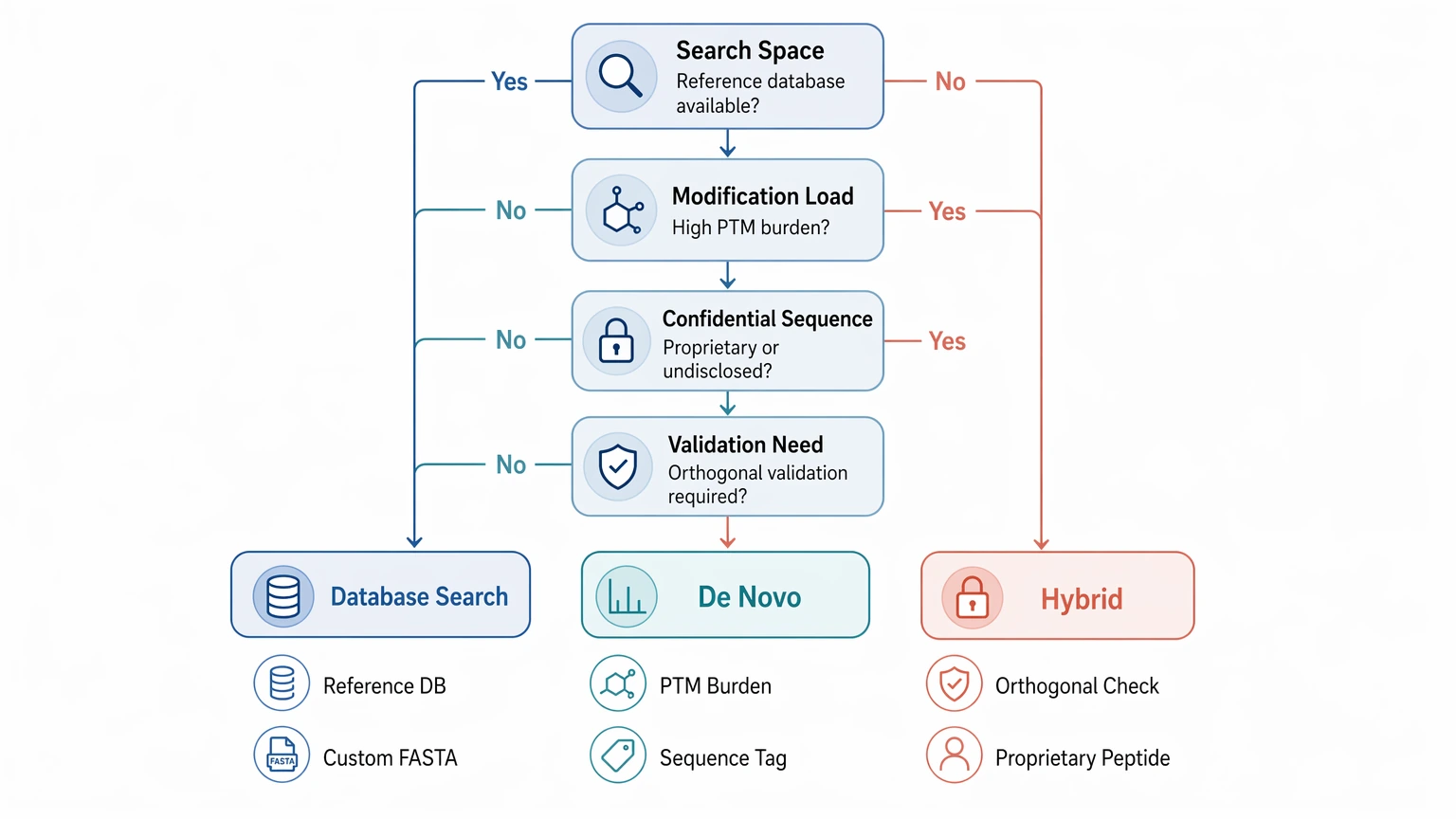
For peptide therapeutics teams, impurity studies, engineered peptide programs, and unknown natural peptide work, the most useful outcome is often not a universal winner between methods, but a bounded interpretation with clear next validation steps. If you need that kind of decision support, contact MtoZ Biolabs to evaluate your project with the sample type, expected peptide class, known or masked sequence constraints, LC-MS/MS files, and the level of sequence confidence required for the next milestone.
FAQ
Can a custom FASTA solve every proprietary peptide problem?
No. A custom FASTA helps only when the candidate sequence space is still meaningfully represented. If important regions are hidden or the true chemistry falls outside the model, de novo evidence is still needed.
Does a high FDR-controlled database result mean the sequence is correct?
It means the match is statistically strong within the search space that was tested. It does not guarantee that the correct peptide was included in that search space.
Is a partial sequence tag useful if it is not a full sequence?
Yes. A sequence tag can rule out many wrong candidates, support custom database design, and guide targeted follow-up, especially in impurity or discovery work.
Are PTM localization and backbone sequence confidence always linked?
Not always. A peptide backbone can be reasonably supported while the exact PTM position remains uncertain, or vice versa.
When should we reacquire data instead of pushing interpretation further?
Reacquisition is often the better next step when spectra are mixed, precursor isolation is poor, or fragment coverage is too sparse to support residue-level confidence.
What information should we prepare before requesting workflow guidance?
Prepare the sample type, approximate purity, expected modifications, any sequence regions that can or cannot be shared, available LC-MS/MS raw files, and the required output format.
How to order?

