





De Novo Peptide Sequencing by Deep Learning: What It Changes in Spectrum Interpretation and Confidence Assessment
- a reliable reference proteome exists
- the goal is routine confirmation
- novelty is not the main question
- spectra are relatively clean
- a short rescue analysis is enough
- detailed confidence mapping is not the main need
- database search fails for structural reasons
- candidate sequence ranking must remain interpretable in incomplete spectra
- residue-level confidence, sequence ambiguity, and orthogonal validation planning will affect the project decision
- partial ion ladders, where only fragments from parts of the peptide are visible
- noncanonical fragmentation patterns, where internal fragments or neutral losses complicate the spectrum
- reference-limited projects, where database search returns no confident peptide-spectrum match
- ranked candidate sequences
- peptide-level confidence
- residue-level confidence or positional uncertainty
- sequence tag support
- precursor mass agreement
- fragment mass error summary
- notes on sequence ambiguity, including leucine/isoleucine ambiguity
- PTM localization status when relevant
- targeted LC-MS/MS to confirm discriminating fragment ions
- synthesis comparison for a leading candidate
- intact mass logic to test compatibility with a larger analyte context
- homolog or assembly review for protein-level interpretation
- complementary characterization when PTM localization remains weak
- Is database search failing because the reference proteome is insufficient, or because the data are weak?
- Does the output show residue-level confidence and sequence ambiguity clearly enough to guide follow-up work?
- Are alternative candidates preserved when the spectrum does not uniquely support one answer?
- Does the report distinguish immediate interpretation from orthogonal validation needs?
When LC-MS/MS data come from unknown, engineered, weakly annotated, or modification-rich peptides, deep learning-based de novo sequencing often gives teams a better decision framework than sending the data through another database search. The gain is not just a higher score. It comes from reconstructing candidate sequences directly from tandem mass spectrometry evidence and showing uncertainty in a form that can actually support the next step.
That distinction matters when a team has to decide whether a proposed sequence is strong enough for synthesis, impurity investigation, biologics characterization, or targeted confirmation. In those settings, the useful comparison is not “AI versus non-AI.” It is whether the workflow improves MS/MS spectrum interpretation and makes peptide-level confidence and residue-level confidence easier to defend.
Quick Decision Guide
Use database search first when:
Use conventional de novo peptide sequencing when:
Use deep learning-based de novo sequencing when:
Core limitation: even strong model output does not remove leucine/isoleucine ambiguity, uncertain PTM localization, or the need to compare the candidate back to the observed MS/MS spectrum.
Why This Comparison Matters
Database search and de novo peptide sequencing answer different questions. A database search asks whether an observed peptide-spectrum match exists inside a defined search space. That remains very effective when the reference proteome fits the sample and the peptide is expected. The weakness shows up when the sequence is novel, highly divergent, truncated, engineered, impurity-related, or modified in a way the search space does not capture well.
De novo peptide sequencing starts from the other end. Rather than asking whether the sequence is in a database, it reconstructs sequence from fragment ions in the MS/MS spectrum. That makes it attractive for unknown peptide identification, but it also creates a harder confidence question: how much of the proposed sequence is directly supported, and where does uncertainty still sit?
Deep learning-based de novo sequencing changed this discussion because it can model fragment patterns beyond simple path tracing between peaks. In practice, that can improve candidate sequence ranking when b ions and y ions are incomplete, when neutral losses complicate interpretation, or when novelty makes database search structurally weak.
How Deep Learning Changes MS/MS Spectrum Interpretation
Conventional de novo tools usually infer residue order from mass differences that match expected fragment ions. The logic is transparent, which is useful, but it can turn brittle when the spectrum has missing cleavage-site evidence, mixed signal, or PTM-shifted fragments. A broken ion ladder often leaves the analyst with short sequence tags or unstable ranking.
Deep learning-based de novo sequencing uses the same physical input data but changes the reconstruction logic. Instead of relying only on explicit mass-difference paths, the model learns statistical relationships between sequence patterns and fragment-ion evidence from large training sets. That can help in three recurring situations:
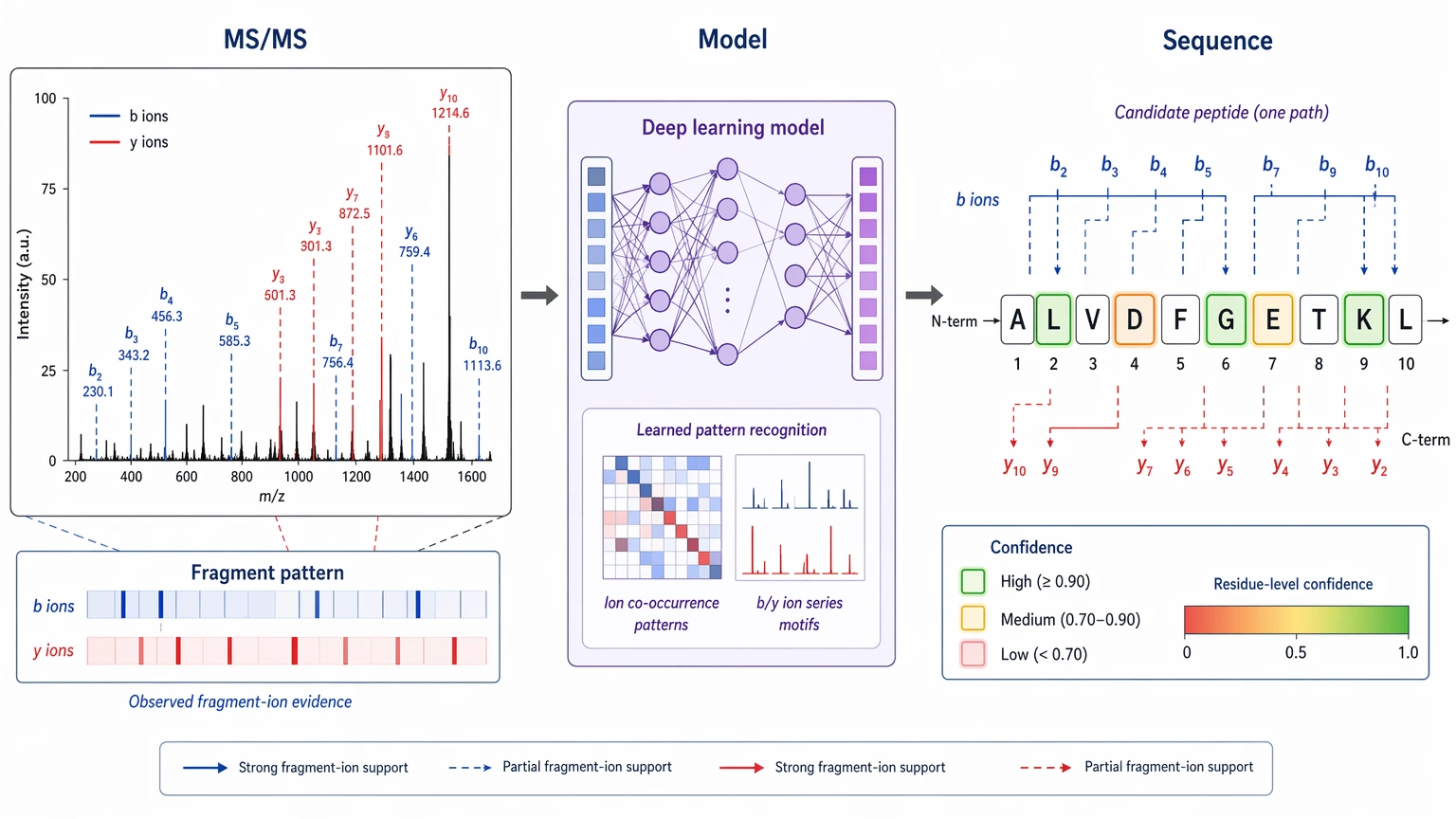
The practical gain is usually better organization of incomplete evidence, not guaranteed full sequence recovery. A model may recover a longer sequence tag, produce a more coherent candidate sequence ranking, or mark local uncertainty more clearly than older tools. Even so, a proposed sequence is still a hypothesis until the observed fragment ions, precursor mass, and fragment mass error behavior line up behind it.
How Confidence Assessment Changes
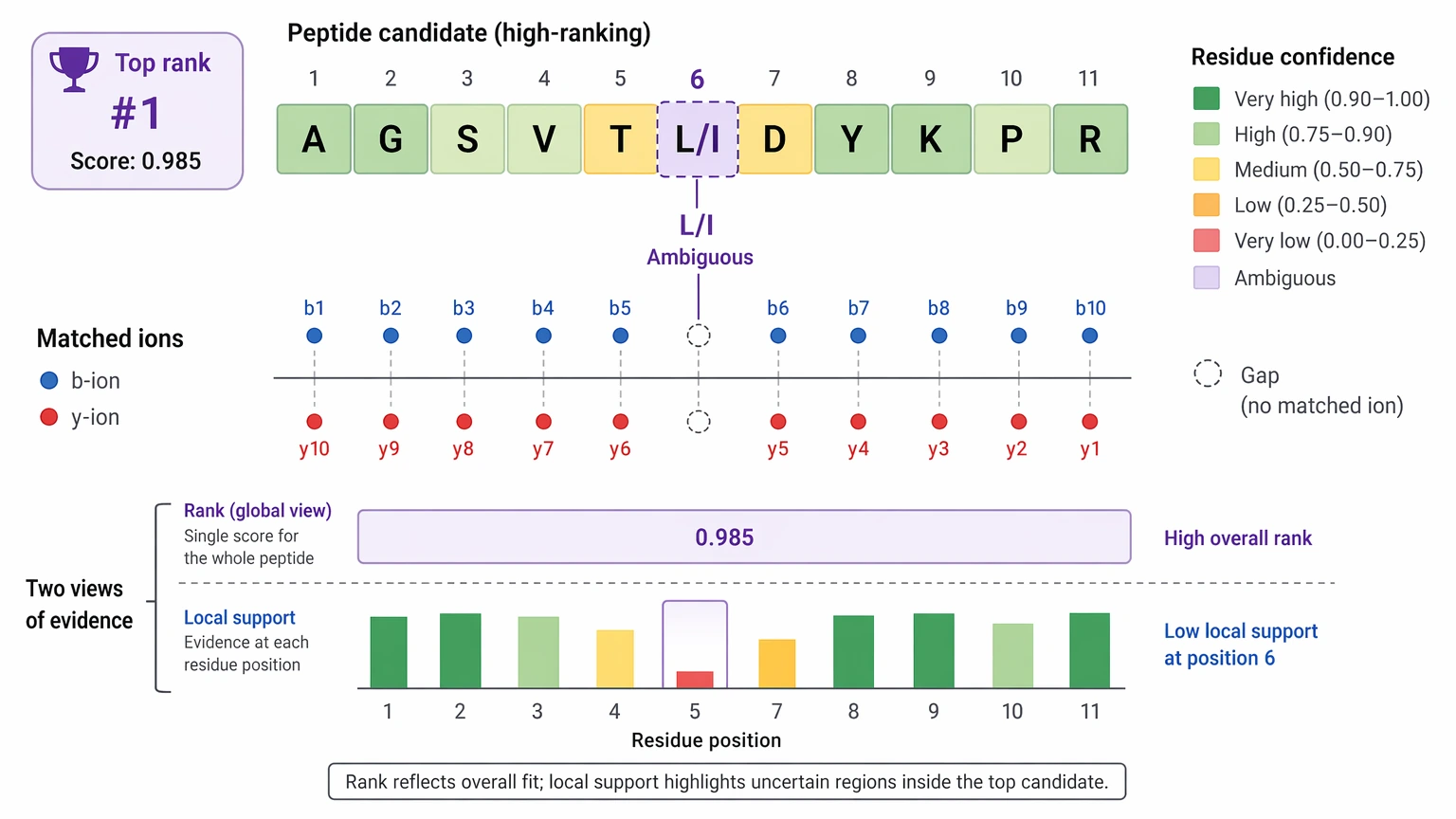
The biggest shift is the confidence structure itself. Older workflows often collapse interpretation into a single peptide-level score. That helps with ranking, but it can hide where the evidence is solid and where it starts to thin out. Deep learning-based de novo sequencing often adds residue-level confidence, and that changes how teams read the result.
A more defensible review usually combines several layers:
| Evidence | What it tells you | Main boundary | Best next check |
|---|---|---|---|
| High residue-level confidence across a continuous region | That segment is strongly supported | Terminal regions may still be weak | Inspect local fragment-ion coverage |
| High peptide-level confidence with precursor mass agreement | The full candidate is globally plausible | Local errors can still be hidden | Review low-confidence positions one by one |
| Long sequence tag supported by b ions and y ions | Backbone order is credible over a useful span | Gaps may contain substitutions or PTMs | Use the tag in targeted confirmation planning |
| Tight fragment mass error across matched peaks | The interpretation is mass-consistent | Mass fit alone does not prove residue order | Compare against alternative candidates |
| Several near-top candidates | Sequence ambiguity is real | Ranking gaps may overstate certainty | Preserve alternatives for validation |
Takeaway: peptide-level confidence and residue-level confidence need to be read together. One helps prioritize the candidate. The other tells you how much validation work still sits ahead.
Service Routes to Consider
For this project scenario, readers usually compare these service routes before requesting a quote or submitting samples.
A high score does not automatically mean the sequence is correct. Sometimes it reflects a strong statistical fit to incomplete evidence. This matters most for isobaric residues. Leucine/isoleucine ambiguity remains a basic MS-only limitation in many workflows, and a top-ranked sequence should not be presented as uniquely resolved if the fragment evidence does not separate those residues.
Where Deep Learning Adds the Most Value
The strongest use cases are specific. They are projects where standard identification logic fails for a known reason.
First, deep learning-based de novo sequencing is often worth evaluating when the search space itself is the bottleneck. That includes species-limited samples, engineered constructs, impurity-related unknowns, and novelty-rich peptide mixtures.
Second, it becomes more useful when confidence needs to be localized rather than averaged. If the project hinges on whether uncertainty sits at one residue, one terminus, or one PTM site, residue-level confidence is more actionable than a single rank score.
Third, it can improve interpretation in difficult but still informative spectra. Examples include partially observed b ions and y ions, spectra affected by neutral losses, and modified peptides where direct database support is weak.
The advantage narrows when the spectra are already simple and the project question is narrow. In clean spectra with clear ion ladders, conventional de novo peptide sequencing may already provide enough information for a limited rescue analysis.
Expected Results and Validation Methods
A good report from deep learning-based de novo sequencing should separate immediate deliverables from follow-up confirmation.
Immediate deliverables should include:
These outputs help a team decide whether the candidate is mainly hypothesis-generating or strong enough to shape the next step.
Follow-up confirmation should be planned separately and may include:
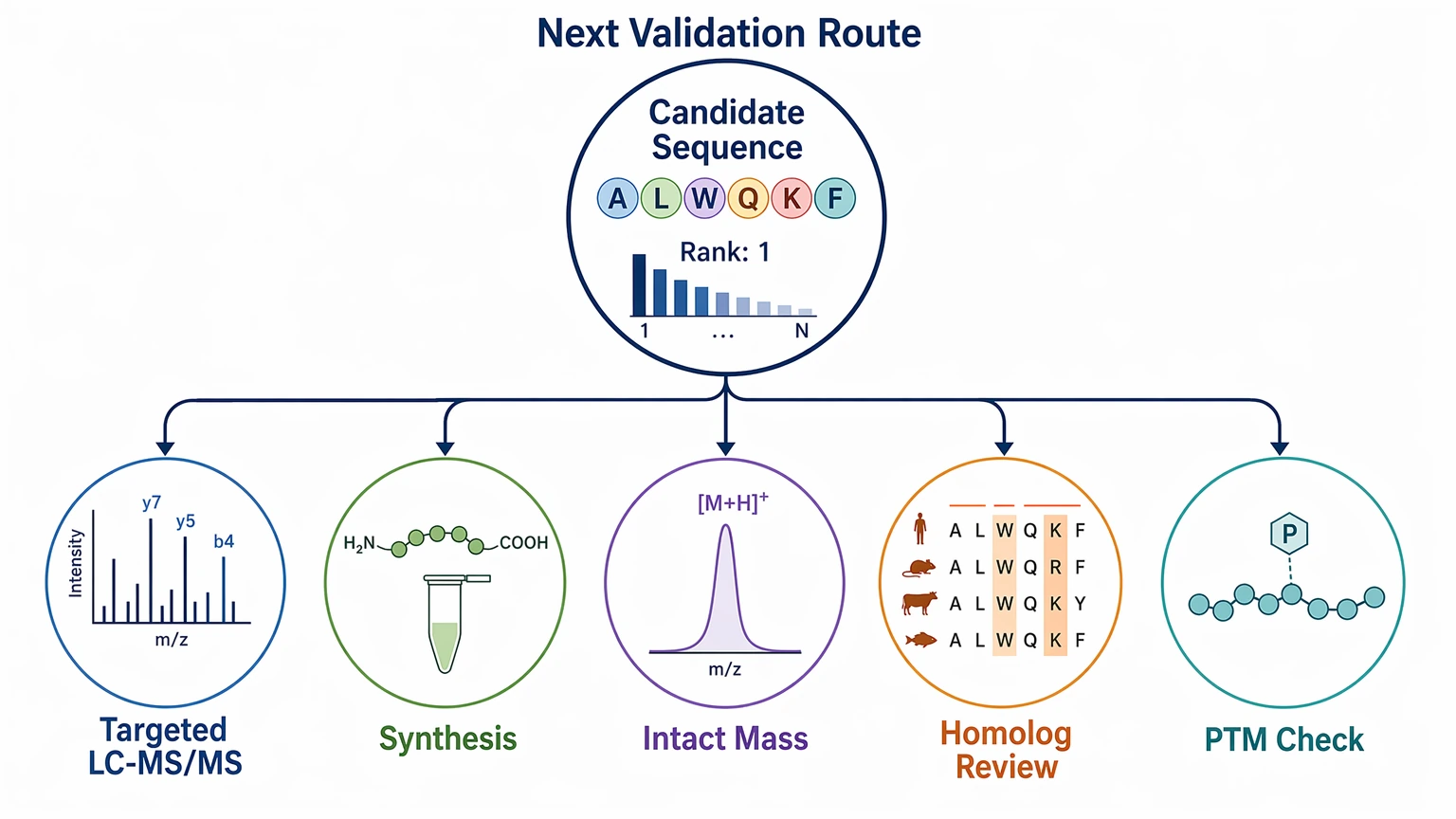
An explicit limitation should stay in the report: for PTM-rich or partially fragmented spectra, MS/MS interpretation may support a ranked candidate sequence without uniquely resolving every residue or modification site.
If you are comparing workflow options before committing resources, MtoZ Biolabs can evaluate your project and help define whether de novo sequencing should serve as the primary route or a validation-guided rescue route; submit your requirements before building downstream assays around a tentative candidate.
Key Cautions and Practical Limits
Deep learning improves interpretation, but it does not remove the main analytical boundaries.
Sample quality or amount limits: low-abundance material, poor precursor isolation purity, or thin fragment-ion richness can reduce sequence confidence regardless of model choice.
Controls and repeat expectations: if a candidate will drive synthesis, impurity assignment, or internal go/no-go decisions, repeat acquisition or targeted confirmation is often justified.
Batch or contamination risk: chimeric spectrum interference, carryover, or mixed precursors can produce plausible but misleading candidate sequences.
Interpretation boundaries: peptide-level de novo evidence should not be stretched into full protein-level certainty without additional assembly logic or orthogonal validation.
When another method is the better next step: if the main problem is poor spectrum quality rather than search-space limitation, reacquisition, cleaner isolation, targeted MS, or a database-centered workflow may be more informative than pushing de novo analysis further.
How to Evaluate a Workflow or Service
A practical review should focus on four questions:
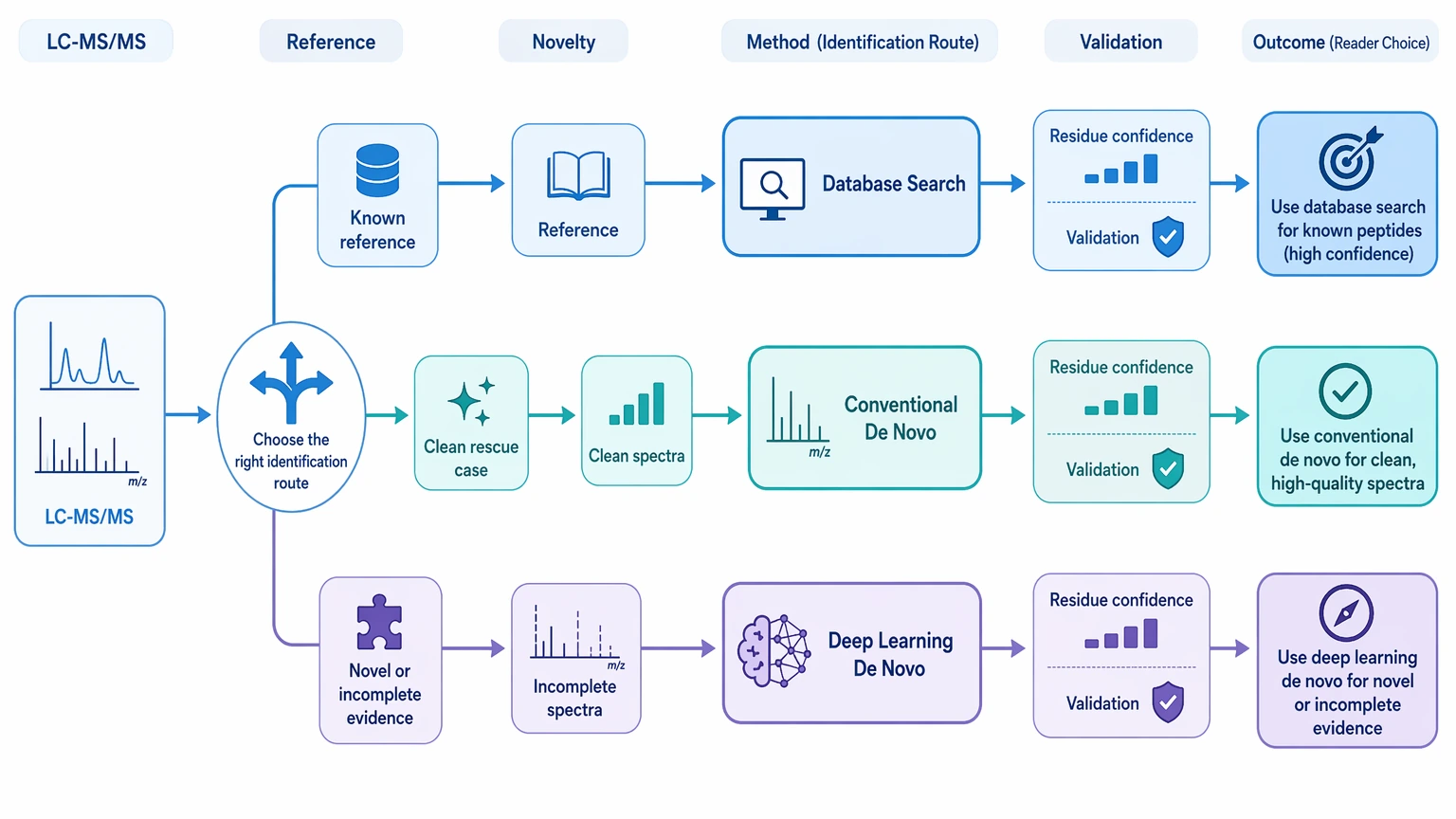
Ask to see how the workflow handles chimeric spectrum risk, PTM localization boundaries, candidate sequence ranking, and fragment-ion support. If the output only gives a top sequence and a score, it may not be enough for a high-consequence decision.
Technical Summary and Consultation Guidance
Deep learning-based de novo sequencing changes de novo peptide sequencing most when LC-MS/MS datasets contain unknown or weakly reference-supported peptides and the team needs a confidence framework they can actually defend, not another pass of database search. Its value comes from better interpretation of incomplete fragment-ion evidence and clearer reporting of residue-level confidence, peptide-level confidence, and sequence ambiguity. It fits novelty-rich, PTM-aware, or rescue-analysis projects well, but it still has firm limits around isobaric residues, chimeric spectra, sparse fragmentation, and protein-level inference. If your project must decide whether a candidate sequence is ready for synthesis, targeted confirmation, or broader characterization, contact MtoZ Biolabs to evaluate your project context, LC-MS/MS data quality, and validation path before treating a de novo call as a final answer.
FAQ
How does training-set bias affect deep learning-based de novo sequencing?
If the model was trained mostly on common peptide classes or fragmentation behaviors, unusual chemistries may be ranked less reliably. That does not make the output unusable, but it does raise the need to compare model confidence with direct fragment-ion evidence.
Should a team run database search and de novo peptide sequencing together?
Often yes. Database search can confirm expected content quickly, while deep learning-based de novo sequencing can rescue unexplained spectra and surface candidate sequences outside the known search space. In many projects, the combination is more informative than forcing one workflow to answer every question.
What should a useful de novo report look like for downstream decision-making?
It should show ranked candidates, residue-level confidence, peptide-level confidence, sequence tag support, precursor mass agreement, fragment mass error behavior, and a clear note on unresolved ambiguities. A single score without spectrum-linked interpretation is usually not enough.
When is reacquiring LC-MS/MS data smarter than pushing interpretation harder?
Reacquisition is often the better next step when precursor isolation purity is poor, fragment coverage is sparse, or the spectrum looks mixed. In those cases, more model sophistication may not make up for weak evidence.
Can deep learning-based de novo sequencing support protein-level conclusions directly?
Only cautiously. Peptide-level de novo evidence can suggest protein identity or sequence deviation, but broader protein reconstruction usually needs peptide integration, homolog review, intact mass context, or other orthogonal validation.
How to order?

