





Antibody Sequencing NGS Quality Metrics: How to Judge Coverage, Error Control, and Paired-Chain Confidence Before Reformatting
- Completeness: Are VH and VL full enough for construct design, including the regions that affect expression-ready sequence assembly?
- Consensus support: Are reported bases supported by read family evidence rather than inflated duplicate counts?
- Biological coherence: Does each chain show a productive rearrangement, an in-frame sequence, and plausible framework region continuity?
- Pair evidence: Is chain pairing direct, linked, or only inferred from sample-level co-occurrence?
- UMI / unique molecular identifier use
- read family construction and family-level consensus generation
- duplicate collapsing logic
- bidirectional support when the workflow supports it
- base-call confidence at amino-acid-changing positions
- agreement across replicate libraries or independent amplifications, when available
- CDR3
- junctional bases
- insertion/deletion-prone regions
- positions shaped by somatic hypermutation
- any site that changes productivity, residue chemistry, or construct design
- coherent V(D)J rearrangement or VJ structure
- in-frame sequence continuity
- absence of a premature stop codon
- plausible CDR lengths
- reasonable germline assignment
- intact framework region organization after translation
- unresolved bases in CDR3
- multiple near-identical contigs with amino-acid consequences
- mixed light-chain signals in a sample expected to be monoclonal
- indirect chain pairing in a mixed input
- missing terminal sequence that blocks construct design
- discordance between replicate calls or orthogonal evidence
An antibody sequencing NGS dataset is ready for recombinant reformatting only when three questions have solid answers: Is the antibody variable region complete enough for construct design, are the key base calls still supported after error suppression, and does the reported heavy chain / VH and light chain / VL pair match the evidence class of the workflow? If any one of those points is still weak, the sequence may still be useful for annotation or early review, but it should not yet be treated as a final cloning input.
That cutoff matters because costs climb fast once a sequence is accepted. A report can show high read counts, a dominant consensus sequence, and a plausible germline assignment and still leave open gaps at CDR3, low-confidence amino-acid-changing positions, mixed light-chain signals, or pairing that was inferred rather than directly linked. Before synthesis or vector design, the practical question is not whether antibody-derived reads exist. It is whether the dataset reaches sequence recovery quality that is good enough for reformatting.
What the QC Decision Actually Means
Here, antibody sequencing NGS means next-generation sequencing used to recover antibody variable region sequences from a hybridoma-derived sample, sorted B-cell material, or another antibody-producing source. The goal is not a general repertoire summary. The real decision is whether the sequencing evidence is already strong enough to support recombinant reformatting or whether orthogonal validation should happen first.
A useful review starts with four technical checks:
Those checks are what separate a sequence that is simply reportable from one that can actually support engineering decisions.
Coverage: More Than Raw Read Depth
Read depth matters only when it is spread across the antibody variable region in a way that supports stable reconstruction. For reformatting, coverage should be reviewed across framework region segments, CDR1, CDR2, CDR3, and the V(D)J rearrangement or VJ junction. If leader-adjacent or terminal sequence is part of the construct strategy, those ends need explicit review too.
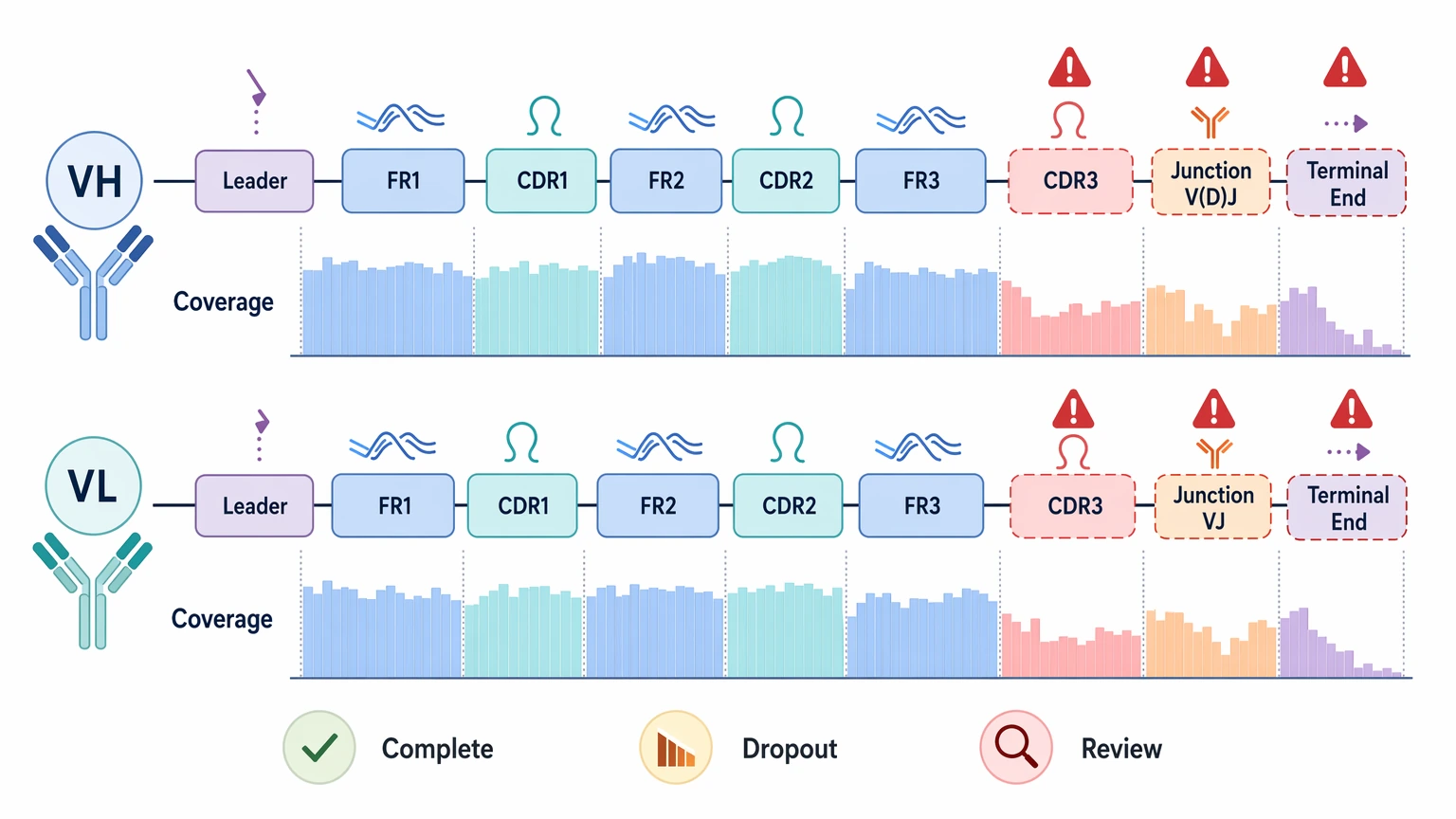
High total reads can still leave you with a weak dataset if support drops out at the exact positions that determine amino acid identity. That is why coverage breadth and coverage uniformity usually tell you more than raw count alone.
| Metric area | Stronger signal for reformatting | Warning sign |
|---|---|---|
| Read depth | Sustained support across VH and VL critical regions | Most reads concentrated in only one portion of the locus |
| Coverage breadth | Full or near-full antibody variable region recovery | Missing terminal sequence or incomplete CDR3-adjacent support |
| Coverage uniformity | Comparable support across framework regions and CDRs | Sharp dropout at junctional positions |
| Consensus structure | One dominant consensus sequence with stable regional support | Multiple near-matching contigs with unresolved amino-acid differences |
| Clonal pattern | Clear clonal dominance in expected monoclonal material | Background diversity inconsistent with the stated sample type |
A hybridoma-derived sample should usually look simpler than bulk B-cell material. If a monoclonal source produces several competing productive rearrangements or mixed VL signals, that is a real warning sign, not a minor cosmetic QC issue.
Error Suppression: Can You Trust the Actual Bases?
In antibody sequence recovery, a single incorrect base can change a CDR residue, introduce a premature stop codon, disrupt a cysteine pattern, or create an artificial glycosylation motif. The damage is often concentrated at a few positions rather than spread across the entire readout, which is why base-level review matters more than broad average quality statements.
The most useful error-control signals include:
A good report should let you tell the difference between a consensus sequence built from multiple independent molecules and one that mostly reflects PCR redundancy. Those are not the same thing. Without a clear error-correction or error-suppression strategy, even a high-count position can still be weak if the support comes from a narrow set of underlying molecules.
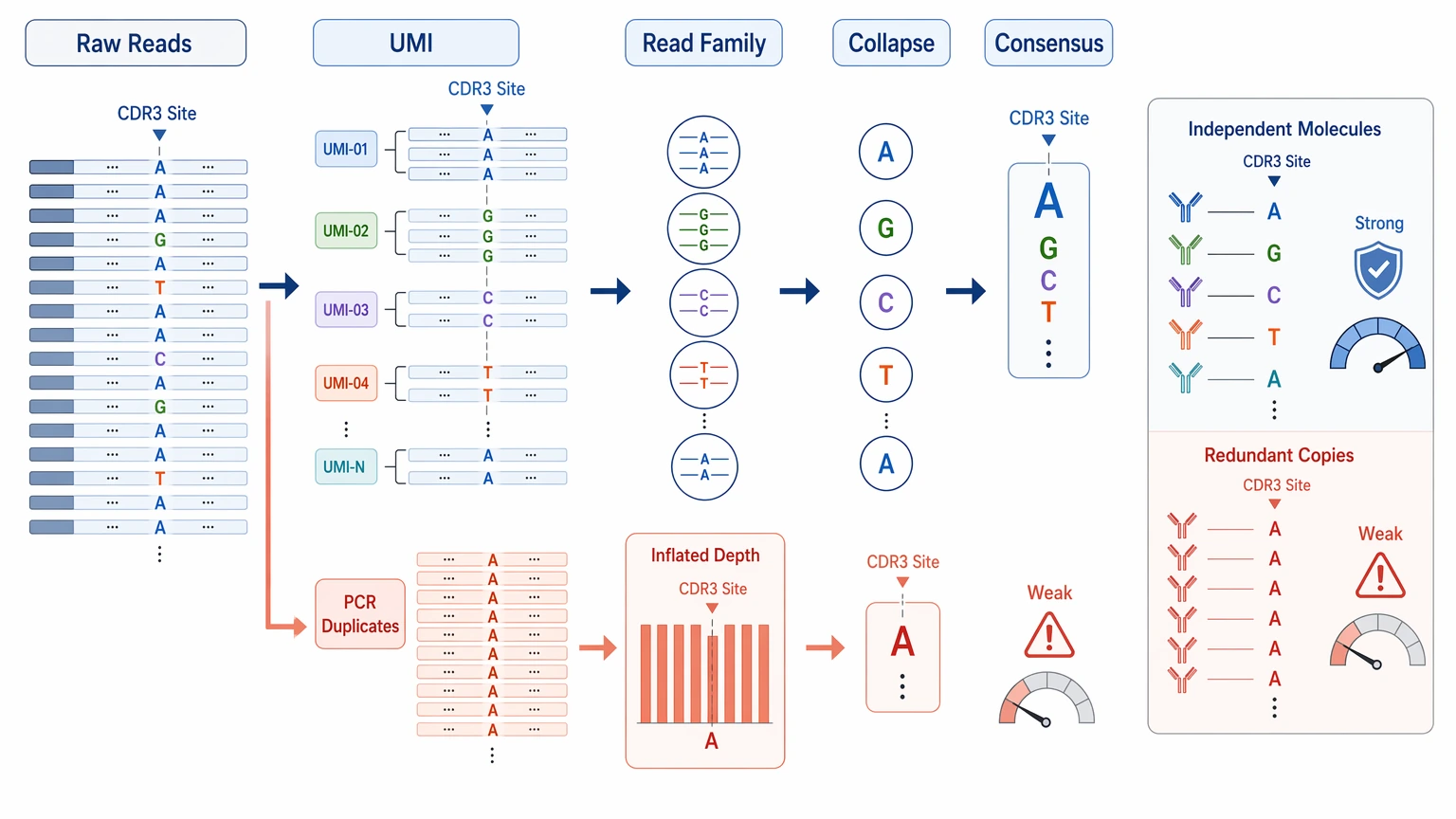
The highest-priority review points are usually:
If your team is deciding whether to order synthesis now, this is often where a closer technical review pays off. When a provider report does not clearly distinguish raw depth from consensus-grade support, submit your requirements for an independent QC review before moving the sequence into reformatting.
Productive Rearrangement and Sequence Plausibility
Coverage and error control still do not tell you whether the recovered chains form biologically coherent antibody sequences. Before accepting a VH or VL call, confirm that each chain shows a productive rearrangement with a plausible antibody architecture.
Key checks include:
Unusual features are not automatically wrong. Somatic hypermutation can push a sequence away from its nearest germline and create residues that look suspicious at first glance. Still, atypical calls should stay internally coherent. A report that combines weak base support with an unusual amino acid change deserves more caution than a report showing that same change with strong read family backing.
Paired-Chain Confidence: How Strong Is the VH/VL Match?
Paired-chain confidence should be judged by evidence class, not by how polished the summary report looks. Seeing one heavy chain and one light chain in the same sample does not, by itself, prove correct chain pairing.
The following framework is practical before recombinant reformatting:
| Pairing evidence class | Confidence level | Interpretation before reformatting |
|---|---|---|
| Single-cell pairing | Highest | Direct heavy/light association from the same cell barcode or compartment |
| Explicit molecular linkage or well-level linkage | High | Strong support if contamination controls are credible |
| Dominant VH and VL from a clean hybridoma-derived sample | Moderate to high | Often workable, but still indirect compared with direct linkage |
| Bulk co-occurrence across repertoire reads | Moderate to low | Suggestive, not decisive |
| Inferred pairing without direct linkage | Lowest | Best treated as provisional until further confirmation |
This distinction changes project decisions. In a clean monoclonal sample, dominant productive VH and VL sequences may be enough to move ahead. In mixed B-cell material, that same evidence would usually justify additional validation first. If your dataset falls into that gray zone, MtoZ Biolabs can evaluate the sample context, pairing evidence, and report structure so you can contact us with a more specific validation question rather than a generic “success or failure” request.
A Practical Review Sequence for Provider Reports
A short review sequence helps keep the decision centered on reformatting readiness rather than generic NGS QC.
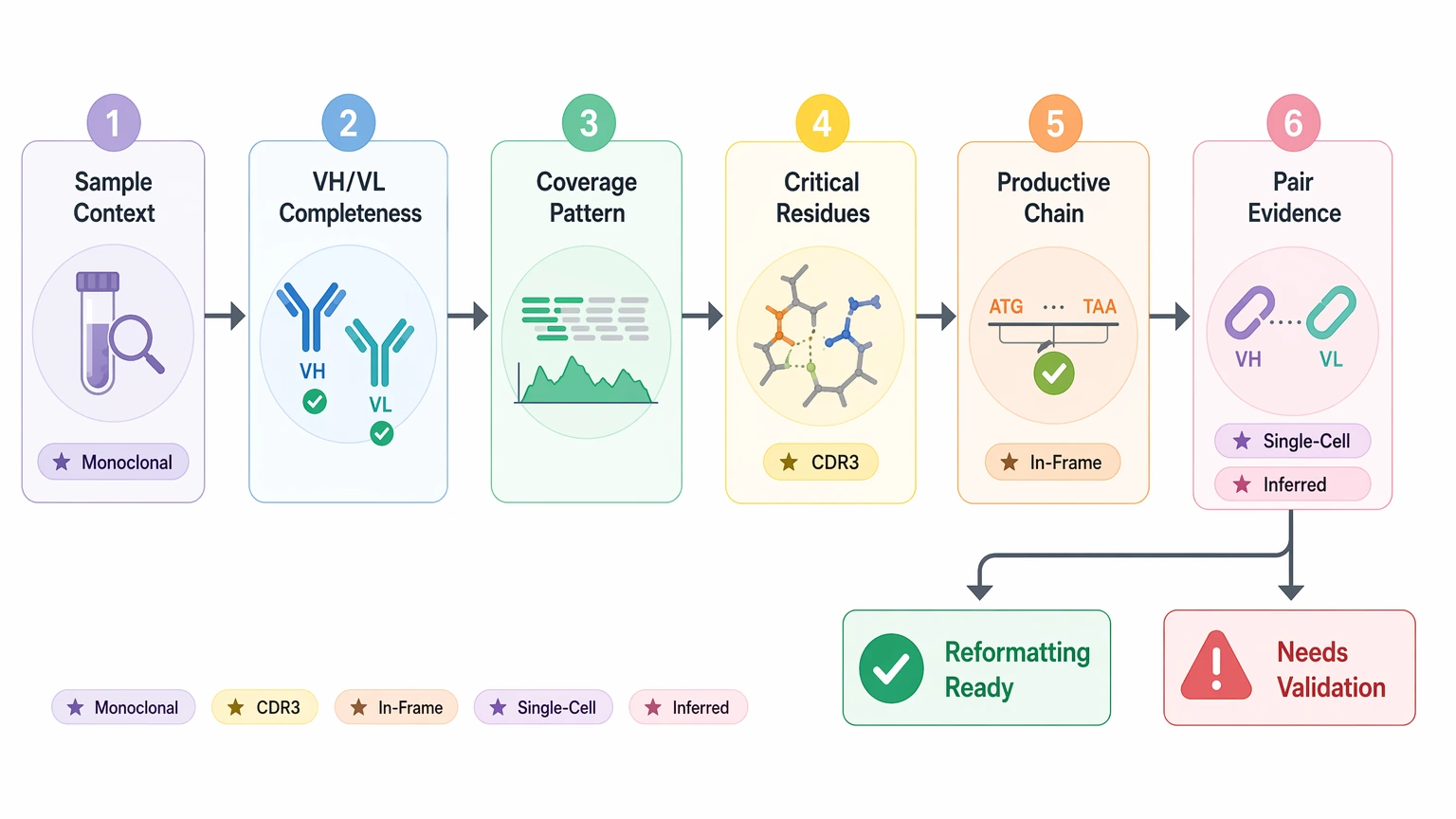
1. Start with sample context
Check whether the biological source is expected to be monoclonal, oligoclonal, or mixed. The same contig pattern means very different things in a hybridoma-derived sample versus bulk B-cell input.
2. Confirm sequence recovery completeness
Review whether VH and VL each cover the antibody variable region needed for design. Partial recovery may still support annotation while remaining insufficient for synthesis-ready construct planning.
3. Compare read depth with coverage breadth and coverage uniformity
Ask whether the sequence is supported where it matters most. Strong framework region support does not make up for weak CDR3 or junction coverage.
4. Inspect the consensus sequence behind critical residues
Look for read family support, duplicate collapsing, and base-call confidence at amino-acid-changing positions. This step matters more than headline read counts.
5. Verify productive rearrangement
Translate the sequence and confirm an in-frame sequence, no premature stop codon, plausible framework continuity, and coherent germline assignment.
6. Classify chain pairing by evidence type
Do not accept “paired” at face value. Ask what experimental feature linked the heavy and light chain and whether the result reflects single-cell pairing, linkage, or inferred pairing.
When Additional Validation Is the Better Choice
A dataset usually needs more work when one of the following conditions appears:
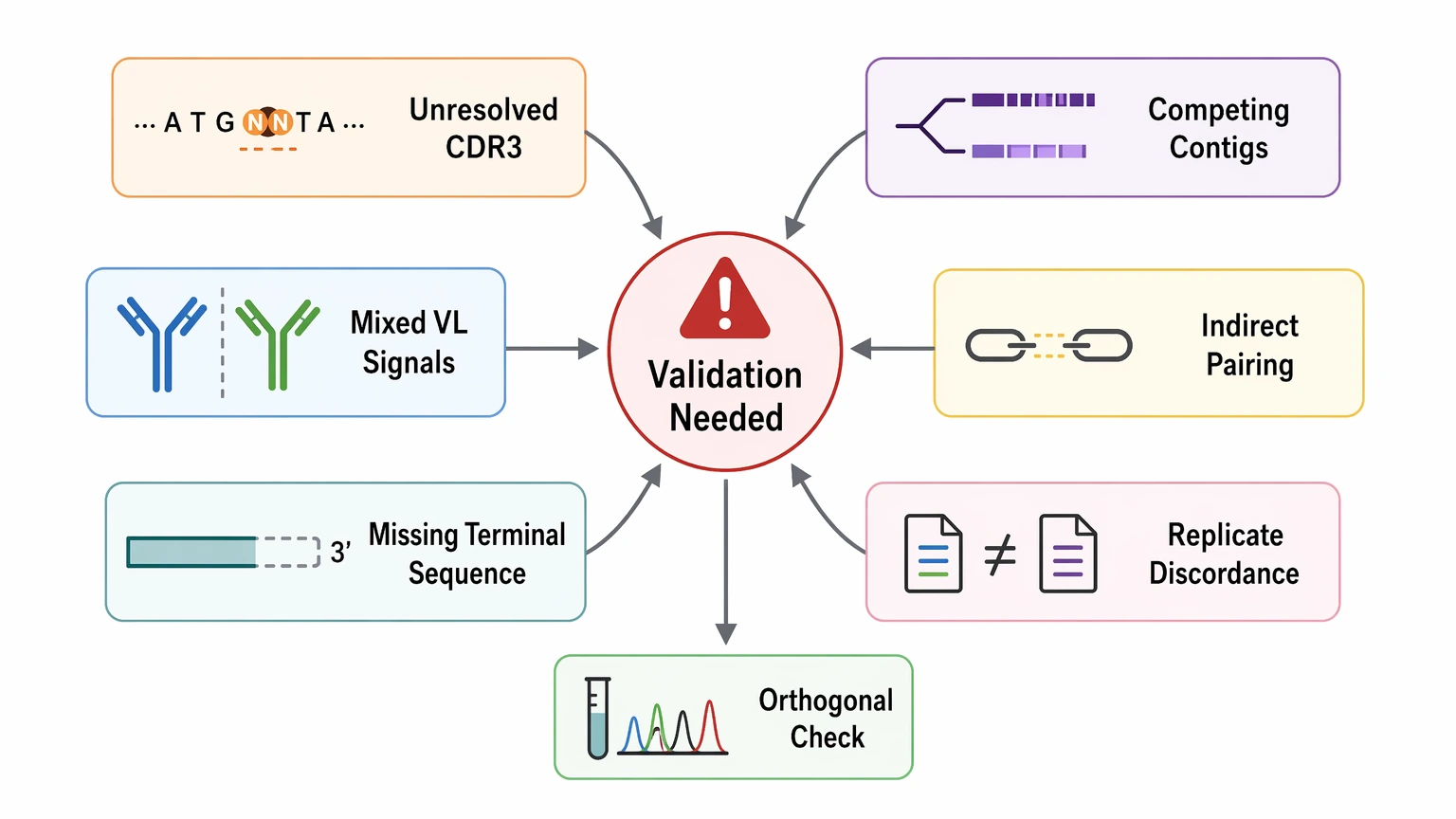
At that stage, the better next step is usually targeted confirmation, not optimistic progression into expression. Orthogonal validation may involve additional amplification, a linkage-aware workflow, or another sequence-confirmation strategy matched to the sample source and project goal.
Conclusion
Before recombinant reformatting, the most useful way to judge antibody sequencing NGS is to ask whether the dataset supports complete sequence recovery, credible error-suppressed base calls, and a chain pairing claim that matches the workflow and sample context. Read depth alone does not answer that question. The stronger indicators are coverage breadth, coverage uniformity, read family-backed consensus sequence support, productive rearrangement, low contig ambiguity, and evidence-classified paired-chain confidence.
For hybridoma-derived projects, mixed B-cell inputs, or provider reports with unresolved CDR3 or pairing questions, a structured technical review can keep weak sequence assumptions from turning into expensive design choices. If your team is deciding whether to move from report review to cloning, contact MtoZ Biolabs to evaluate your project, discuss the sample and data context, and define whether the current evidence is reformatting-ready or better treated as provisional pending orthogonal validation.
FAQ
Can a sequence be acceptable for annotation but still weak for recombinant reformatting?
Yes. Annotation may tolerate partial terminal coverage or limited uncertainty at noncritical positions. Recombinant reformatting usually requires a more complete and stable sequence because synthesis, cloning, and expression planning assume the reported VH and VL are functionally usable inputs.
Why do mixed light-chain signals matter more than minor heavy-chain background in some reports?
A mixed VL pattern can directly affect construct identity because light-chain ambiguity changes the expressed antibody pair. Minor heavy-chain background may be less disruptive if one productive VH clearly dominates and the extra signals are weak, nonproductive, or biologically implausible.
Does a strong germline assignment reduce concern about a weak CDR3 base call?
No. Germline assignment supports the broader identity of the rearrangement, but CDR3 often contains the most consequential sequence uncertainty. A confident germline label cannot rescue a poorly supported junctional residue that changes amino acid identity.
Should amino acid translation be reviewed even when the provider already labels the sequence as productive?
Yes. Provider labels are useful, but direct translation review can expose frame shifts, borderline indel calls, suspicious cysteine changes, or stop codons that were masked by an overly simplified summary.
What makes a bulk-sample pairing claim stronger, even without single-cell pairing?
Confidence improves when the sample context supports low complexity, one VH and one VL dominate by a wide margin, both chains are productive, and contamination controls are clear. Even then, the result remains weaker than direct linkage and should be described as inferred pairing rather than proven pairing.
Is recombinant expression failure always evidence that the NGS sequence was wrong?
No. Expression can fail for reasons unrelated to the recovered variable-region sequence, such as vector design, codon optimization, expression format, or construct assembly issues. Sequence confidence and expression outcome should be reviewed together, not treated as interchangeable proof.
How to order?

