





Antibody Sequencing by Mass Spectrometry vs Gene-Based Recovery: Which Route Fits Your Project
- Protein only available: start with antibody sequencing by mass spectrometry
- Hybridoma, B cells, RNA, cDNA, or plasmid available: start with gene-based recovery
- Need to know the actual product state in hand: favor LC-MS/MS-based protein evidence
- Need a coding sequence for redevelopment with lower residue ambiguity: favor gene-based recovery
- Both evidence types available and reuse risk is high: consider a combined strategy
When the starting material is incomplete, the most useful first step is to match the route to the strongest evidence you actually have. Start with antibody sequencing by mass spectrometry when purified antibody, formulated drug product, or other protein-only material is the only dependable source. Start with gene-based recovery when you have a viable hybridoma, B-cell source, RNA, cDNA, plasmid, or a traceable sequence record and the goal is to recover an expressible coding sequence.
Quick decision block
These routes overlap, but they answer different questions. Antibody sequencing by mass spectrometry reconstructs sequence from LC-MS/MS and tandem mass spectrometry peptide evidence. Gene-based recovery reads nucleic acid or linked sequence records more directly. The better fit depends on sample reality, the required sequence confidence in the variable region and CDR, the burden of post-translational modification (PTM), and how much orthogonal validation the project can support.
Where teams usually make this decision
This choice usually comes up during project review, not basic method selection. Teams inherit a legacy monoclonal antibody, receive only a vial of purified IgG, lose plasmid records, or need to reproduce an older reagent for redevelopment or comparability work. At that point, sample conservation matters, and so does the risk of choosing a route that cannot answer the real project question.
A protein-only sample will not move in a gene-centered workflow without a credible cellular or nucleic acid source. On the other side, teams sometimes expect de novo sequencing to settle more than the protein evidence can really support, especially when sequence coverage is uneven, peptide overlap is sparse, PTM-heavy regions interfere, or multiple antibody populations are present. The real question is not which platform sounds stronger. It is which workflow matches the evidence in hand and the decision that comes next.
What each route starts from
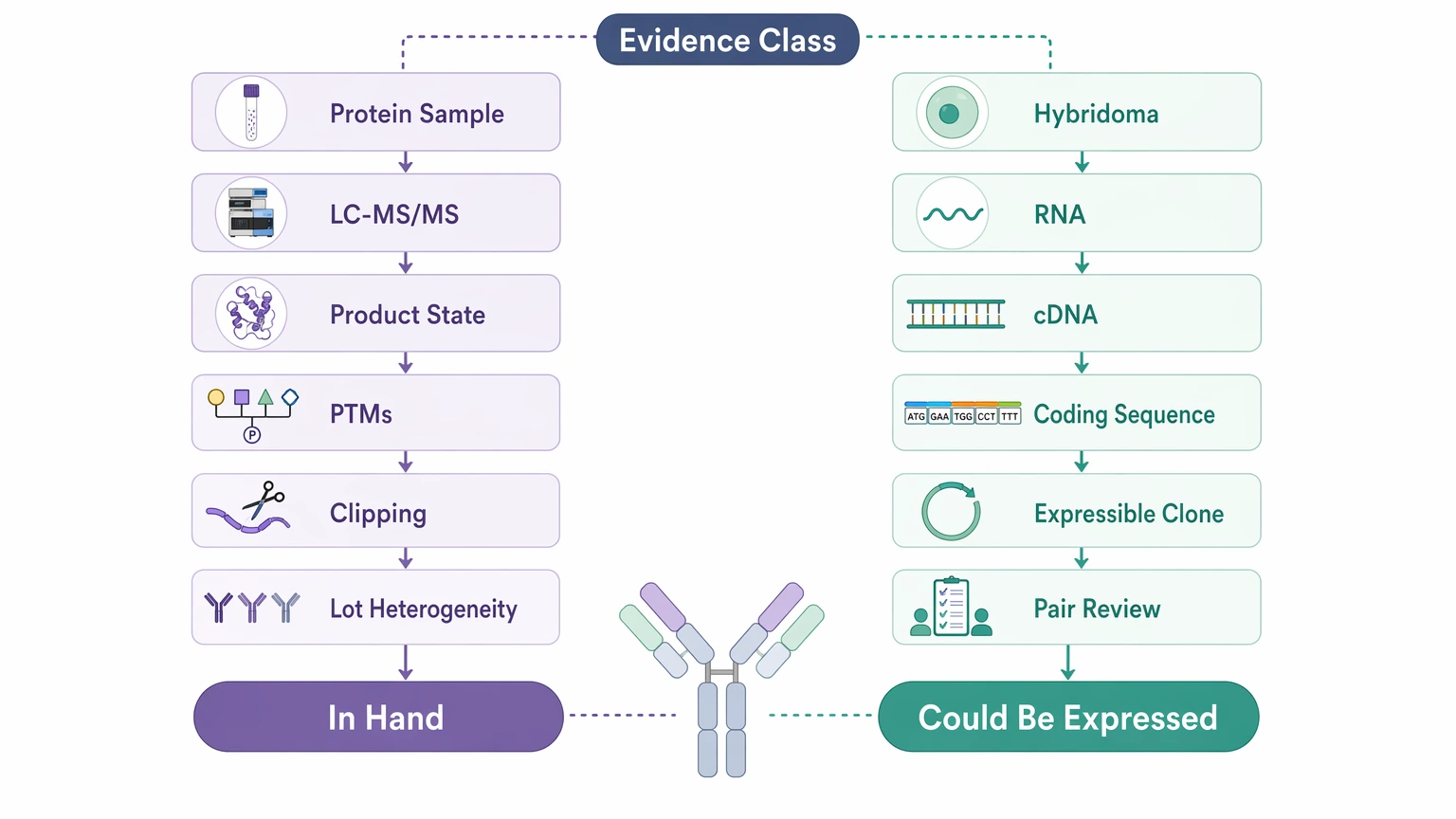
The main difference is the evidence class.
Antibody sequencing by mass spectrometry starts from the protein. A standard bottom-up sequencing workflow uses digestion, peptide mapping, and MS/MS interpretation to recover heavy chain and light chain sequence information across the variable region and constant region. When no cellular source remains, this is often the only realistic starting point.
Gene-based recovery starts from biological source material or records tied to it. That can include a hybridoma, sorted B cells, PBMC-derived B-cell material, RNA, cDNA, or a plasmid archive. In the right setting, B-cell receptor sequence recovery or cDNA / RNA-based recovery can provide coding sequences more directly than protein inference.
That distinction matters because gene evidence and protein evidence support different decisions. A gene sequence tells you what could be expressed. Protein evidence is better for showing what is physically in the sample, including clipping, PTMs, and lot-specific heterogeneity.
Head-to-head comparison by project reality
Use the table below to choose the first route before you spend scarce sample or trigger follow-up work.
| Scenario | Better starting route | Main risk | Usual follow-up |
|---|---|---|---|
| Only purified antibody is available | Antibody sequencing by mass spectrometry | Coverage gaps in the variable region | Peptide remapping and intact mass review |
| Formulated antibody drug product only | Antibody sequencing by mass spectrometry after formulation assessment | Excipients and PTM burden complicate spectra | Cleanup planning plus orthogonal validation |
| Hybridoma cells are available | Gene-based recovery | Recovered transcripts still need correct chain pairing | Pair review and protein-level confirmation if reuse is planned |
| B-cell source or RNA is available | B-cell receptor sequence recovery or cDNA / RNA-based recovery | Pairing confidence may be limited by workflow design | Recombinant expression confirmation |
| Plasmid or archival sequence records exist | Gene-based recovery first | Records may not match the current material in hand | Spot-check with peptide mapping or intact mass |
| Protein and gene-source material are both available | Combined strategy | More planning and cross-confirmation effort | Compare gene-derived and protein-derived evidence |
The table works best as triage. Start with the strongest evidence source, then add a second layer only if the remaining uncertainty could block redevelopment or comparison.
Service Routes to Consider
For this project scenario, readers usually compare these service routes before requesting a quote or submitting samples.
When antibody sequencing by mass spectrometry is the better starting route
This route is strongest when the protein is the only trustworthy evidence, or when the actual product state matters. That includes legacy antibody rescue, undocumented reagents, and lots with formulation history or suspected processing changes.
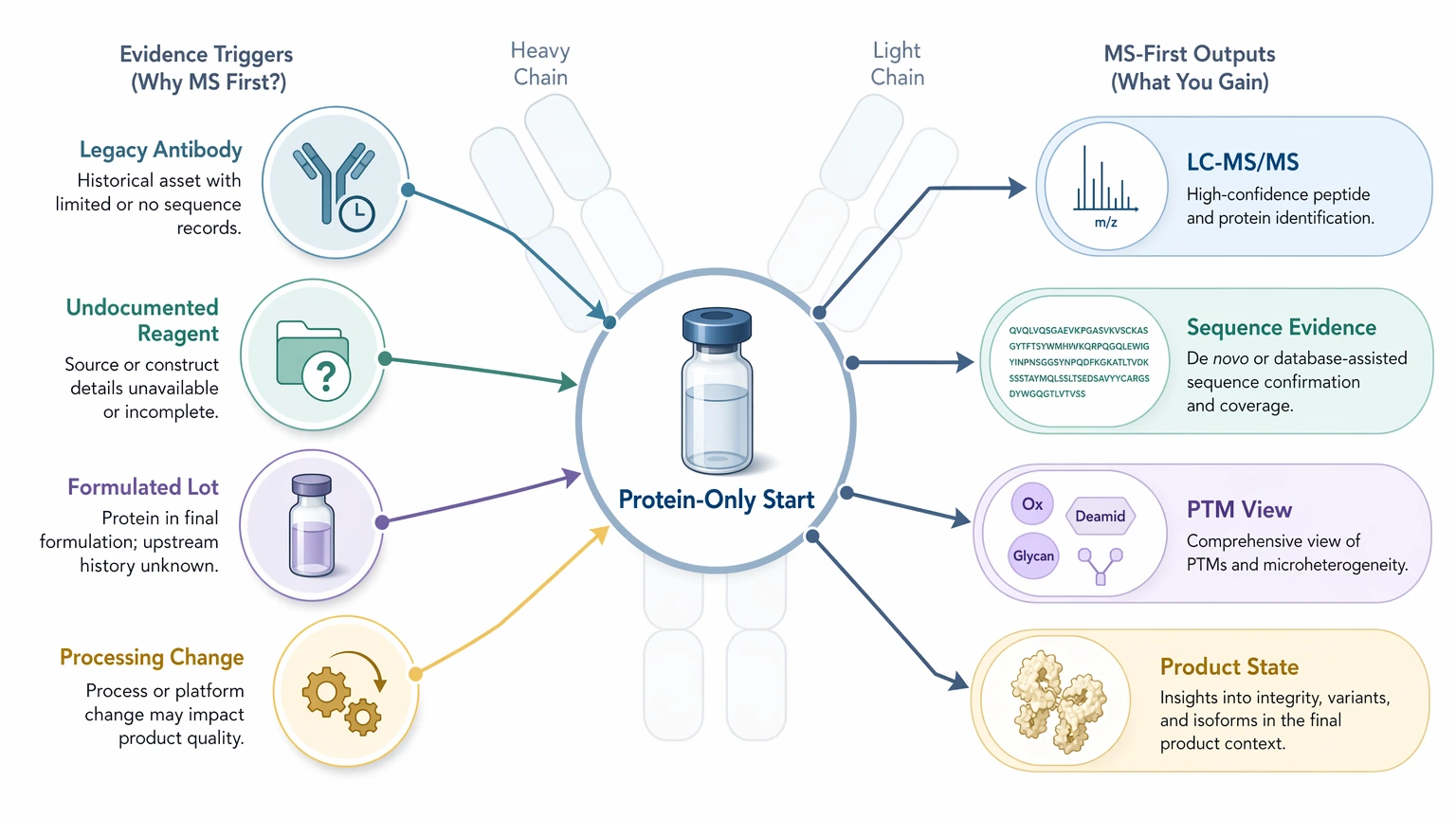
A well-designed LC-MS/MS workflow can recover large parts of the heavy chain and light chain, often with useful support across the variable region and many complementarity-determining region (CDR) peptides. It can also expose protein-level features such as glycosylation, oxidation, deamidation, pyroglutamate formation, clipping, and disulfide bond complexity. If the team needs to know what is really in the vial, gene recovery alone will not settle that.
The limits still matter. Standard MS workflows do not directly resolve every isoleucine/leucine ambiguity. Some regions fragment poorly, some CDR segments have thin overlap support, and mixed populations or formulation background can complicate chain pairing and local interpretation. In practice, the output is often a candidate sequence with evidence annotations and unresolved positions, not an unconditional final truth statement. One limitation should always be stated plainly: MS/MS-based sequence interpretation can leave uncertain residues or local alternatives when peptide evidence is incomplete, PTMs obscure fragmentation, or database-supported matching is weak.
When gene-based recovery is the better starting route
Gene-based recovery is usually more direct when a viable source is available and clearly linked to the antibody of interest. If the main goal is to rebuild an expression construct, compare a historic clone, or restore an archived reagent for research use, this route often reduces residue-level ambiguity earlier in the project.
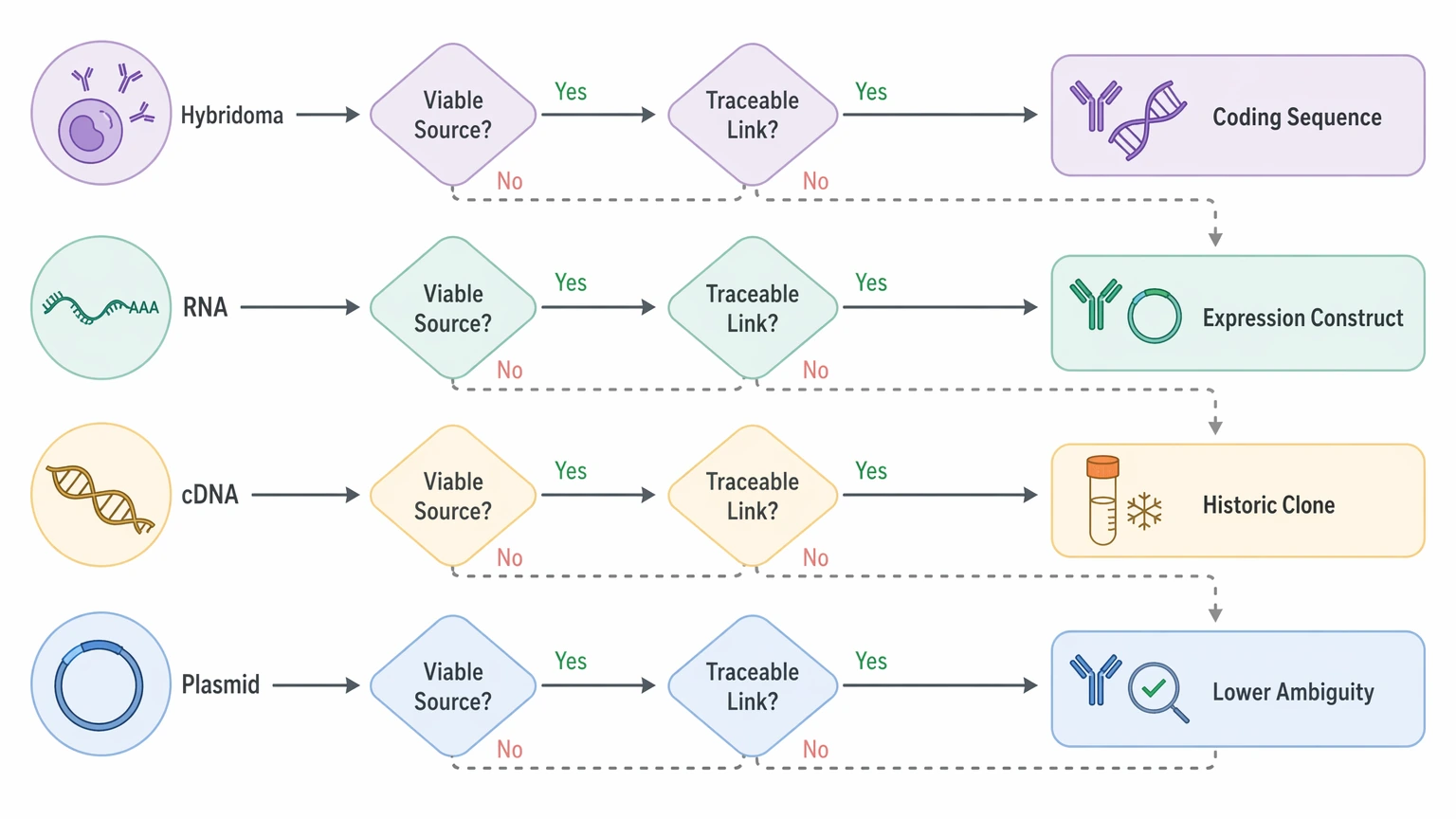
That advantage is clearest when the material includes a preserved hybridoma, clean RNA, cDNA, or a traceable plasmid. In those cases, the project may reach candidate coding sequences faster than a protein-led reconstruction, especially if the protein sample is heavily formulated or chemically modified.
Gene evidence also has limits. Chain pairing is not guaranteed unless the workflow preserves or reconstructs the correct heavy-light pairing logic. A recovered transcript may not be the productive pair the project needs. Likewise, an archived plasmid or cDNA record may reflect the intended construct rather than the exact protein lot now in hand. When the decision depends on the real sample rather than the historical source, gene recovery alone can leave the key question open.
Scenario-based route selection
A few common scenarios make the choice easier.
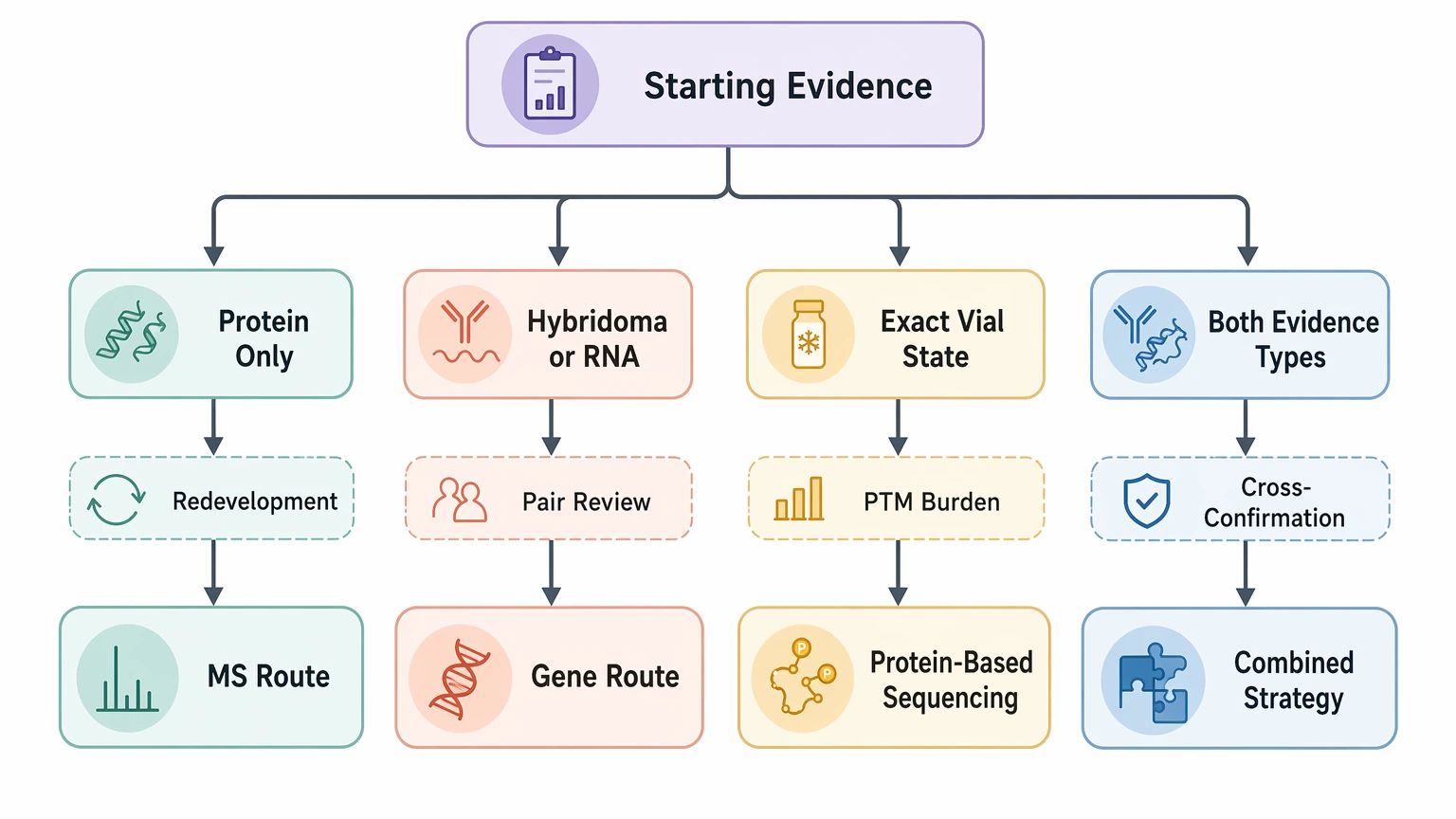
If the project is legacy antibody rescue with no source cells, start with antibody sequencing by mass spectrometry. There is little value in forcing a nucleic-acid route when protein is the only direct evidence.
If a hybridoma or credible RNA source is available and the goal is recombinant redevelopment, start with gene-based recovery. This is usually the cleaner path to an expression-ready sequence, provided pairing is reviewed.
If the team must confirm the exact product present in a vial, including clipping or PTM burden, start with protein-based sequencing even if partial records exist.
If both evidence classes are available and the output will support a sensitive comparison, redevelopment decision, or legal review, the extra planning of a combined route is often justified. If you need to compare these routes against your actual sample constraints, submit your requirements to MtoZ Biolabs to evaluate your project around sample type, likely ambiguity points, and the most defensible confirmation plan.
Expected results and validation methods
The first deliverable from either route is usually a starting evidence package, not the end of the project.
Immediate deliverables from antibody sequencing by mass spectrometry often include candidate heavy chain and light chain sequences, peptide-level evidence, sequence coverage summaries, confidence annotations in the variable region, notes on unresolved residues, and supporting intact mass or peptide mapping results where available.
Immediate deliverables from gene-based recovery often include candidate coding sequences, transcript or record traceability notes, and an initial view of pairing confidence.
Follow-up confirmation differs by route. After MS-based recovery, teams often use peptide remapping against the proposed sequence, targeted LC-MS/MS review of uncertain sites, and comparison of expected versus observed intact mass. After gene-based recovery, confirmation usually centers on recombinant expression confirmation, protein-level spot checks, and review of whether the expressed material matches the historical or physical sample closely enough for the intended use.
Key cautions and practical limits
Several constraints should shape the decision before work begins.
First, sample quality and amount still set the boundaries. Low protein amount, excipient background, degraded RNA, or poorly preserved cells can narrow what either workflow can recover.
Second, controls and repeat expectations should be defined early. Some samples justify repeat digests, targeted confirmation, or a second evidence layer before a team treats a sequence as redevelopment-ready.
Third, batch effects and contamination risk can distort interpretation. Mixed antibody populations, carrier proteins, host-cell background, or sample swaps can create misleading evidence if chain assignment logic is weak.
Fourth, interpretation has boundaries. MS-based outputs may retain unresolved sites because of isoleucine/leucine ambiguity, PTM interference, or incomplete peptide support. Gene-based outputs may still leave doubt about pairing or about whether the recovered source truly matches the product in hand.
Finally, another next step may sometimes be better. If the project needs a direct coding sequence more than product-state insight, gene recovery may be the better first move. If neither evidence class is strong enough on its own, outside feasibility support can prevent unnecessary sample loss. For route triage, validation planning, or a combined workflow review, you can contact MtoZ Biolabs to discuss the study with the sample context and expected deliverable already defined.
Comparison summary and consultation guidance
For antibody recovery projects, the practical rule is simple: choose the route that matches the strongest evidence source and the decision the team actually needs to make. Antibody sequencing by mass spectrometry fits protein-only, legacy, PTM-aware, and product-in-hand questions. Gene-based recovery fits cases with viable source material and a strong need for an expressible coding sequence with fewer residue-level interpretation limits. A combined workflow makes sense when redevelopment or comparability work cannot absorb uncertainty in CDR assignment, pairing, or lot-to-record matching.
For projects such as legacy reagent rescue, redevelopment support, or verification of an archived antibody, the most defensible next step is a technical summary of the available evidence, a route choice matched to the real sample, and a validation plan sized to the reuse risk. Before requesting a route assessment, gather the sample type, amount, formulation details, source history, desired deliverable, and whether orthogonal validation or recombinant expression confirmation will be required.
FAQ
Can peptide mapping alone replace de novo sequencing in an undocumented antibody project?
Usually not. Peptide mapping works best when you already have a candidate sequence to test. In an undocumented antibody project, it more often supports reconstruction than replaces de novo sequencing.
Does gene-based recovery prove that the current antibody vial matches the recovered sequence?
No. It can recover a plausible coding sequence, but it does not directly prove that the vial contains the same processed or modified protein form.
What makes CDR interpretation harder than constant-region interpretation in MS-based work?
CDR peptides are more sequence-variable, often have less overlap, and may contain residues that are harder to assign confidently from fragmentation evidence alone.
If both heavy chain and light chain transcripts are detected, is pairing solved?
Not automatically. Detecting both chains does not confirm that they belong to the correct functional pair unless the recovery workflow preserves or reconstructs pairing logic.
When is a combined protein-plus-gene strategy worth the extra effort?
It is most useful when both evidence classes are available and the project cannot tolerate uncertainty in sequence identity, chain pairing, or match to the material in hand.
What information should a feasibility request include before route selection?
Include sample type, approximate amount, formulation or buffer details, whether hybridoma or RNA exists, whether archived records are available, and the expected endpoint such as sequence archive, redevelopment, or product comparison.
How to order?

