





Single Cell Antibody Sequencing for Paired Heavy-Light Chain Discovery: Planning Samples and Outputs
- the sample is limited, so the team cannot afford several exploratory runs
- the target clone is rare, making broad unsorted input less efficient
- the group wants candidate-ready outputs, not only raw sequencing files
- the sample is cryopreserved, and post-thaw cell condition is uncertain
- the project owner has not defined what “usable output” means for handoff
- choose broader input when the response is not expected to be extremely rare and you want wider survey value
- choose subset sorting when the likely compartment is already known
- choose antigen-focused enrichment when recovering a small number of relevant pairs matters more than describing the full background repertoire
- cell barcode or cell-level identifier
- paired VH and VL sequence
- variable region boundaries
- CDR3 nucleotide and amino acid sequence
- V and J gene calls with germline assignment
- isotype information where applicable
- clonotype group labels
- abundance or supporting cell counts
- candidate flags for clone prioritization
- decide how candidates will be ranked
- identify which paired clones move forward
- confirm sequence integrity if additional review is needed
- produce selected antibodies by recombinant expression
- test binding and other properties with orthogonal assays
- the starting material is purified antibody rather than viable cells
- a hybridoma line already exists and direct sequencing is simpler
- the team only needs repertoire-level trends without preserved pairing
- cell condition is too poor to support credible single-cell capture
- the target population is so diffuse that biological enrichment strategy remains undefined
Single cell antibody sequencing is usually the better choice when the goal is not broad repertoire description, but recovery of paired heavy-light chain sequences from individual B cells for downstream candidate selection. If your team needs native pairing preserved, wants to rank clones at the cell level, and plans to move selected VH and VL pairs into recombinant antibody expression, this workflow is often more useful than bulk B-cell receptor (BCR) profiling.
For planning, start with two practical questions. First, can your material enter the assay as a clean single-cell suspension with enough viable cells for cell-resolved capture? Second, what output package will the downstream team actually use? A project that returns assembled paired sequences, sequence annotation, clonotype grouping, CDR3 calls, and germline assignment is usually more valuable for discovery than a generic sequencing report.
Where project decisions usually stall
Most teams get to this point after the biology has already been narrowed. They may have PBMCs, sorted memory B cells, plasmablasts, or a limited pool of antigen-specific B cells from immunization, infection, or binder screening. At that stage, the question is not whether B-cell sequencing is possible. The real question is whether the selected workflow will preserve the link between the immunoglobulin heavy chain and immunoglobulin light chain from the same cell.
That difference matters. Bulk V(D)J sequencing can describe chain usage, clonal expansion, and repertoire structure, but it does not inherently keep the same-cell heavy-light relationship intact. For antibody discovery, losing that relationship can slow clone recovery because the downstream expression team still needs biologically plausible VH and VL pairings, not separate heavy- and light-chain lists.
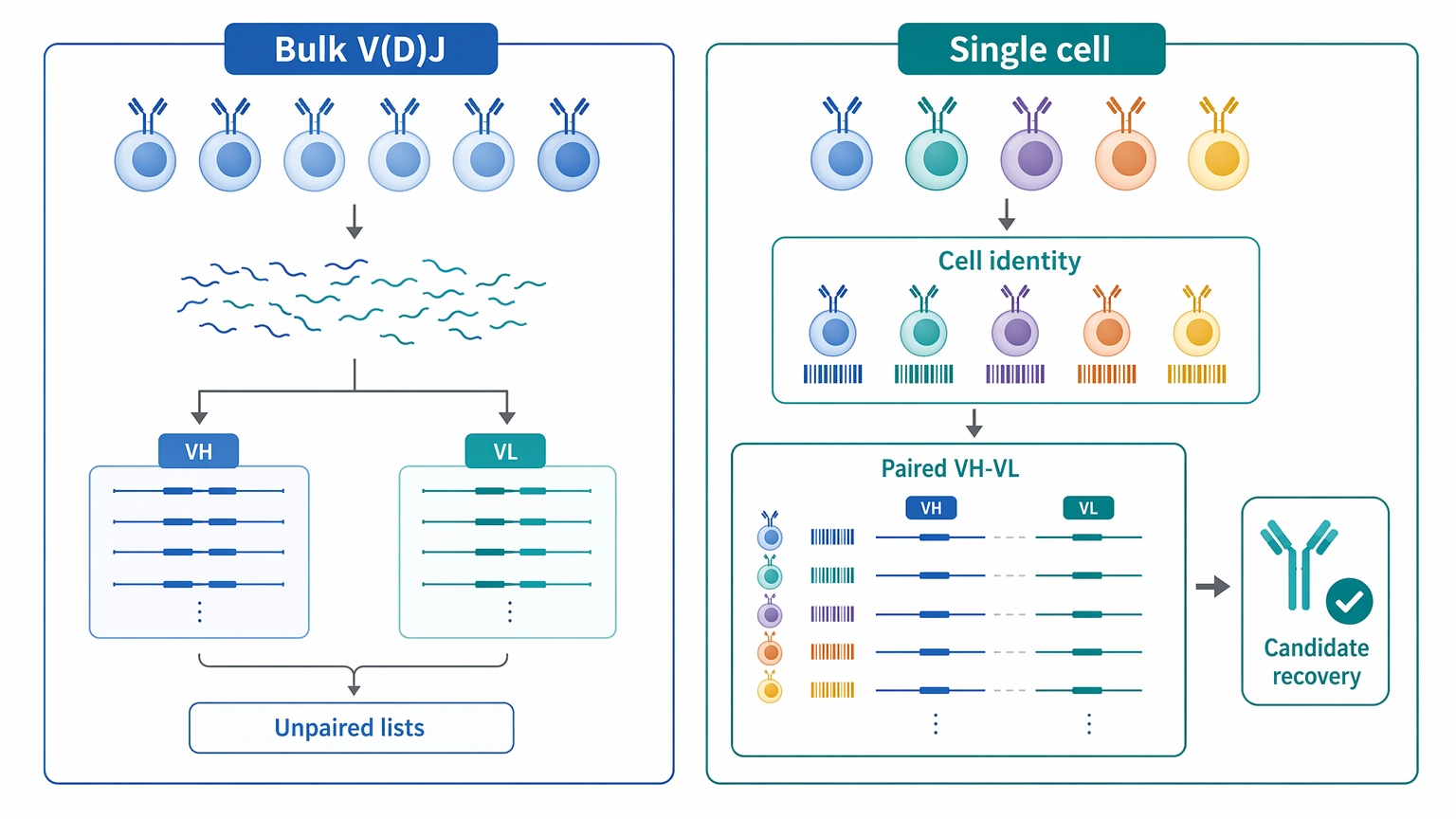
The main planning challenges are usually straightforward:
A sequencing run can succeed technically and still miss the actual decision need. For example, a report centered on repertoire trends may be informative, but not enough for clone prioritization if it does not return paired VH and VL candidates in a usable format.
The planning risks that matter most
For this decision, four cause categories deserve the most attention.
1. Sample condition can limit cell-resolved recovery
Single cell antibody sequencing depends on intact cells rather than extracted nucleic acid alone. Poor viability, excess debris, aggregation, or post-thaw damage can reduce effective B-cell capture and raise doublet risk. Because the value of the workflow is same-cell pairing, weak cell condition affects more than yield. It also affects confidence in the pairing result itself.
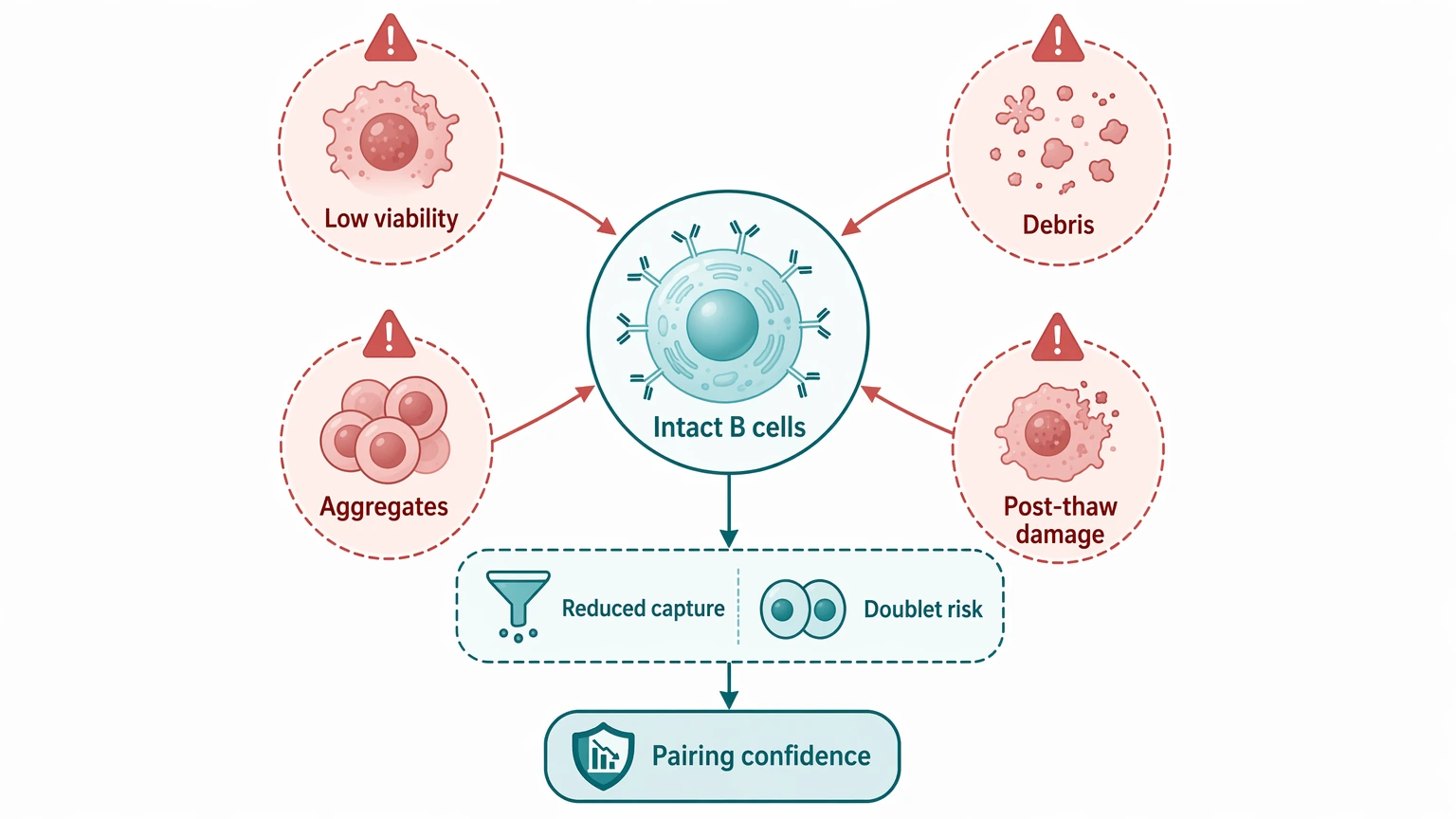
2. The input population may be too broad for the discovery goal
If the objective is to find uncommon but relevant binders, total PBMC loading can dilute the search space. Enrichment or targeted cell sorting often does more for rare-clone recovery than simply ordering more sequencing output. This is especially true when the biology already points to memory B cells, plasmablasts, or antigen-specific B cells as the most likely source of candidates.
3. Output planning may be too vague
Many teams ask for “paired sequencing data” without defining whether they need raw FASTQ files, assembled paired sequences, candidate tables, or expression-ready exports. That ambiguity creates delays that are easy to avoid. The sequencing provider may deliver valid data, but the internal handoff still slows down if annotation fields, file structure, or clonotype organization do not match the next step.
4. Sequence data may be asked to answer functional questions
Paired sequence recovery can support clone selection, lineage review, and re-expression planning. It does not show binding, affinity, neutralization, or developability on its own. A good project plan separates what sequencing can establish from what still needs to be confirmed by expression and orthogonal testing.
A project-planning path for deciding fit
Step 1: Define the decision the dataset must support
Write the downstream use in one direct sentence. A strong version sounds like this: “We need paired VH and VL sequences from a selected B-cell population so we can rank candidates for recombinant expression.” That is much clearer than “we want immune repertoire data.”
This step helps separate use cases. If the real need is chain frequency, repertoire diversity, or general clonal tracking, bulk BCR approaches may be enough. If the real need is candidate antibody recovery with preserved native pairing, single cell antibody sequencing is the better fit.
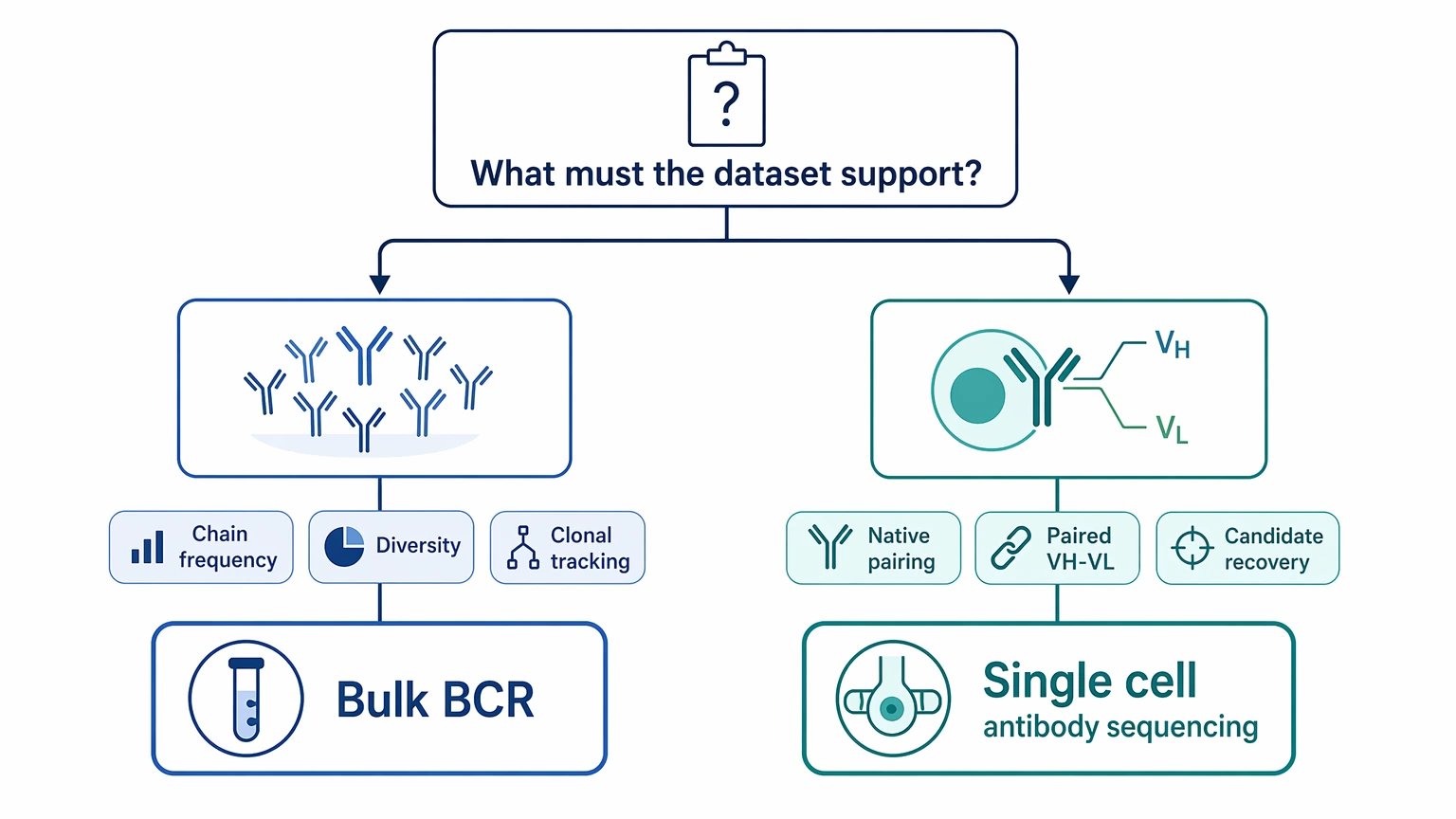
Step 2: Match the starting material to the workflow
Practical sample sources often include PBMCs, enriched total B cells, sorted memory B cells, sorted plasmablasts, or sorted antigen-specific B cells. Fresh material can make handling easier, but cryopreserved material may still work when post-thaw integrity remains acceptable.
Use this table to define the sample conversation before submission:
| Sample source | Discovery value | Main planning check | Common limitation |
|---|---|---|---|
| PBMCs | Moderate to high | B-cell frequency and sort strategy | Large background cell population |
| Enriched total B cells | High | Purity and debris control | Mixed B-cell states |
| Memory B cells | High | Relevance to response biology | May miss short-term effector clones |
| Plasmablasts | High | Collection timing | Narrow sampling window |
| Antigen-specific B cells | Very high | Sort specificity and yield | Low input and sort stress |
Useful readiness signals include acceptable viability, manageable debris, low aggregation, and a realistic path to a clean single-cell suspension. Fit is weaker when the starting material is poorly viable, biologically undefined, or no longer cell based, such as purified antibody or existing hybridoma material.
Step 3: Decide whether enrichment is worth the added complexity
For rare clone discovery, pre-selection can materially change the chance of finding useful paired candidates. If the expected binders represent a small fraction of total B cells, targeted input often improves relevance more than broad loading.
The logic is simple:
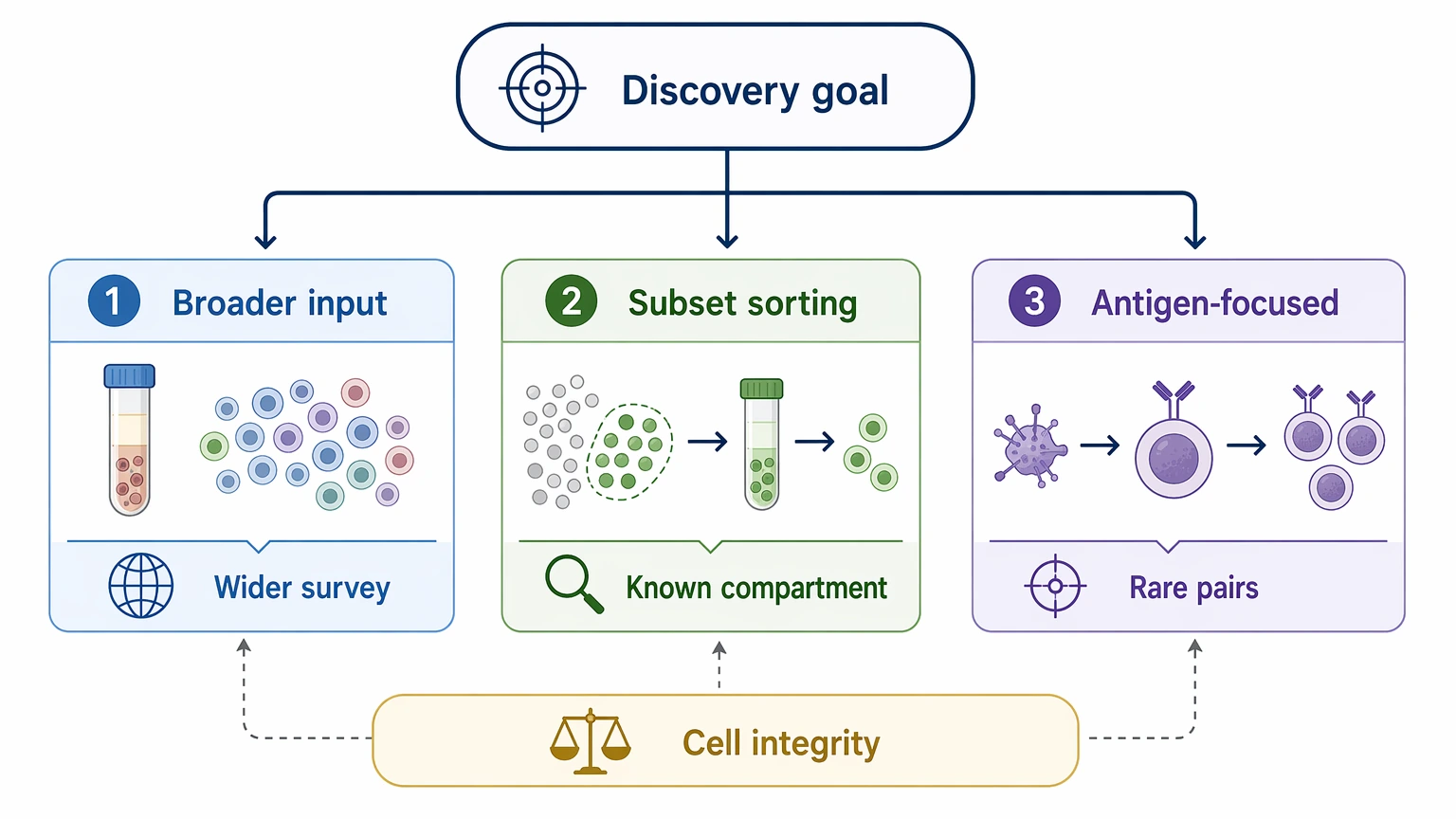
This is also the point where teams should think about doublets and cell stress. A more selective sort is not automatically better if it compromises single-cell integrity. The best plan balances biological focus with recoverable, well-separated cells.
If your group is deciding between unsorted PBMCs, subset selection, or antigen-guided enrichment, submit your requirements and evaluate your project with MtoZ Biolabs around the planned cell source, sorting logic, and paired-chain output needs before launch.
Step 4: Define the output package before sequencing begins
Output design should reflect the handoff, not just the assay. For antibody discovery, the most useful deliverables usually include both data access and candidate-oriented interpretation.
| Output type | Why it matters | Typical downstream use |
|---|---|---|
| Raw FASTQ | Supports independent review | Internal bioinformatics reanalysis |
| Assembled paired VH and VL sequences | Preserves native pairing in usable form | Candidate transfer |
| Sequence annotation table | Speeds filtering and review | Clone prioritization |
| Clonotype summary | Adds expansion and family context | Ranking related candidates |
| CDR3 and germline assignment | Supports lineage and feature review | Candidate triage |
| Expression-ready export | Simplifies next-step transfer | Recombinant antibody expression planning |
Useful fields often include:
If recombinant re-expression is expected immediately after sequencing, ask in advance how the paired sequences will be formatted for synthesis review or construct design. That requirement should shape the report from the start rather than being added after delivery.
Step 5: Set interpretation boundaries and the validation path
A good paired-sequence dataset can tell you which B cells yielded linked VH and VL sequences, which candidates cluster into related clonotypes, and which clones appear expanded or unusual by sequence features. Those are strong discovery signals. They are not functional proof.
For that reason, the validation path should be defined at the same time as the sequencing scope:
That sequence keeps the project grounded. Sequencing helps you recover and organize candidates; follow-up assays determine whether those candidates perform as hoped.
When this workflow is not the best fit
Single cell antibody sequencing is not the automatic answer for every antibody recovery project. Another route may make more sense when:
In those situations, the planning question shifts from “How do we preserve native pairing?” to “What starting material can give the most reliable sequence recovery for this project stage?”
Conclusion
Single cell antibody sequencing is most useful when your project needs paired heavy-light chain recovery from viable B cells in a form that can support clone prioritization and later recombinant antibody expression. The best outcomes usually come from matching sample source, cell condition, enrichment strategy, and output definition to that specific decision instead of ordering a general repertoire study. For teams working with PBMCs, subset-sorted B cells, or rare antigen-focused populations, contact us at MtoZ Biolabs to discuss the sample scenario, define the paired-sequence deliverables, and evaluate the project before submission.
FAQ
Do we need antigen-specific sorting before starting?
Not always. If the response is already reasonably enriched in the available compartment, a broader B-cell input may still be informative. Antigen-specific sorting becomes more attractive when the candidate population is expected to be very rare and the project cannot support screening many unrelated clonotypes.
Is isotype information enough to rank candidates for expression?
Usually not by itself. Isotype context can help interpret B-cell state, but expression decisions are usually stronger when isotype is reviewed together with paired VH and VL sequence quality, clonotype context, CDR3 features, and the biological source of the cells.
Should the downstream team receive nucleotide sequences, amino acid sequences, or both?
Both are often useful. Nucleotide sequences support synthesis planning and traceability, while amino acid sequences help engineering and sequence review. If the expression team is external or separate from the sequencing team, clarify the preferred format before the project starts.
Can single-cell paired-chain data distinguish expanded lineages from independent rare clones?
Often yes, but only within sequence-based interpretation. Clonotype structure, CDR3 relationships, and shared V-gene usage can suggest lineage-related groups, while distinct paired sequences may point to independent clones. Functional convergence still needs experimental confirmation.
What is the most common reporting gap that slows handoff after sequencing?
A report that contains paired sequences but lacks organized annotation tables. Teams often lose time when they have to reconstruct clonotype grouping, germline calls, or variable-region boundaries before deciding what to express first.
How should we compare two sample collection time points for paired discovery?
Plan the comparison before submission. If time point A and time point B are both important, keep collection, thaw, sorting, and library-preparation logic as consistent as possible. Otherwise, apparent differences in clone recovery may reflect processing variation as much as biology.
How to order?

