





Protein Sequencing Methods Compared: Choosing the Right Strategy for Your Research
- Choose the Edman route when only terminal confirmation is required, the sample is purified, and the terminus is accessible.
- Choose database-assisted tandem MS when a trustworthy reference exists and the goal is to confirm that the sample matches the expected sequence.
- Choose de novo sequencing when the sequence is unknown, proprietary, engineered, or likely to differ from available references.
- Choose full-length assembly when the deliverable is a broader sequence map with overlap support across the protein backbone.
Introduction
Protein sequence is often assumed to be known before an experiment begins. In practice, many projects still need direct sequence evidence. A purified gel band may not match any database entry. A recombinant product may differ from the intended construct. An antibody may lack complete genetic records. A biologic may require terminal confirmation before release documentation. In these settings, researchers must choose among several protein sequencing methods rather than defaulting to the workflow used for routine identification.
The decision is not about finding one universally best technique. Edman degradation, database-assisted MS mapping, de novo MS sequencing, and full-length assembly each answer different questions. Terminal methods confirm starts or ends efficiently. Database-assisted workflows confirm identity when a reliable reference exists. De novo sequencing recovers sequence when references are missing or untrustworthy. Full-length assembly combines overlapping peptide evidence for broader coverage. Selecting the wrong path can waste sample, produce false confidence, or leave the biologically important region unreported.
Related Services
| Research Need | Recommended Service Direction |
|---|---|
| MS-based protein sequence confirmation | |
| Full-length sequence recovery | |
| Classical terminal sequencing | / |
| Sequence without reliable database match | |
| Unknown or poorly annotated proteins |
When the best method path is unclear, MtoZ Biolabs can help evaluate whether terminal sequencing, database-assisted mapping, de novo assembly, or full-length MS workflow best matches the sample, coverage goal, and reporting standard.
When Researchers Face This Decision
The comparison usually appears at a specific project gate. A team may need only N-terminal confirmation for a purified product. Another group may need to verify that an expressed protein matches an expected construct. A discovery lab may need sequence from a protein with no reliable reference. An antibody program may need variable-region coverage when transcript data are incomplete. A comparability study may need residue-level proof for a variant-containing region.
In each case, the practical question is the same: what level of sequence evidence does the project require, and can that evidence be obtained with a reference-based or reference-free workflow? Answering that question early prevents mismatched digestion design, unnecessary MS depth, and late-stage disputes about whether the reported sequence is fit for its intended use.
Four Comparison Dimensions That Matter Most
A useful comparison should focus on decision-relevant differences rather than generic method descriptions. Four dimensions matter most across protein sequencing methods: coverage depth, reference dependence, sample suitability, and reporting certainty.
1. Coverage Depth
Terminal sequencing reads a limited number of residues from one end. Database-assisted tandem MS can map many peptides when a reference is valid. De novo sequencing can recover novel or divergent segments. Full-length assembly aims for broad backbone coverage through overlapping peptides and often multiple protease digests.
2. Reference Dependence
The Edman route does not require a database. Database-assisted workflows depend on a correct reference entry. De novo sequencing is designed for cases where the true sequence cannot be assumed. Full-length assembly may combine database matching and de novo interpretation depending on project design.
3. Sample Suitability
Terminal methods work best on purified proteins or peptides with accessible termini. MS-based routes can analyze digested material from gels, recombinant products, and enriched bands, but performance drops in highly complex mixtures unless the target is fractionated. Blocked termini, heavy glycosylation, and low input all influence method choice.
4. Reporting Certainty
Terminal reads are direct but limited in length. Database matching is efficient when the reference is correct. De novo and full-length routes can provide stronger sequence proof for unknown or engineered proteins, but they usually require manual spectrum review, overlap validation, and clear ambiguity labeling.
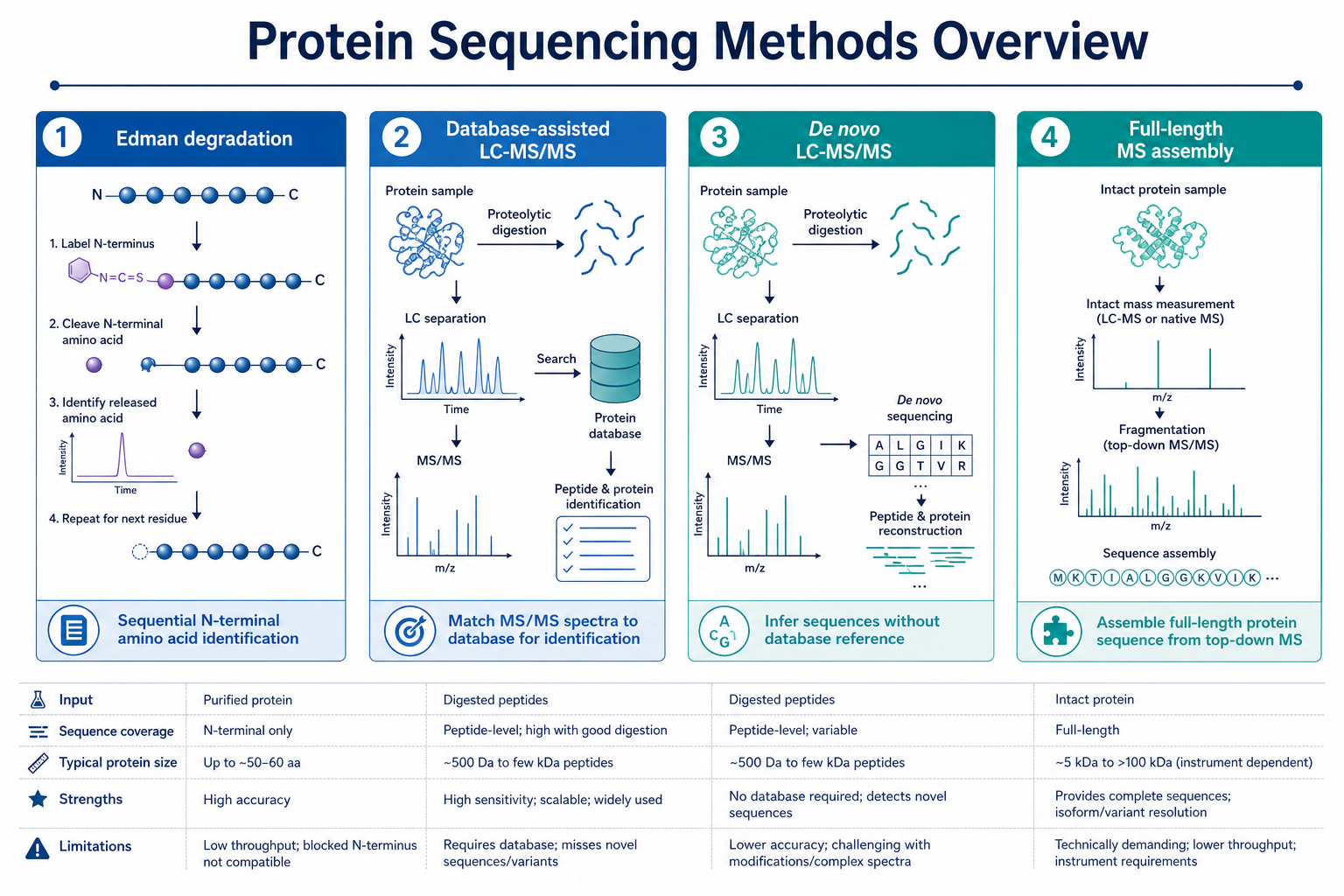
Figure 1. Major sequencing approaches differ in coverage depth, reference use, and reporting logic
Researchers should compare methods by the biological decision behind the project, not by instrument brand or generic lab habit. A method that works well for proteome-wide identification may be the wrong choice when the deliverable is a sequence map for cloning or regulatory review.
Method Comparison at a Glance
The table below summarizes how the major approaches differ on the dimensions most teams use during project planning. It is a planning guide, not a substitute for sample-specific feasibility review.
| Method | Typical Coverage | Reference Needed? | Best Sample Type | Main Strength | Main Limitation |
|---|---|---|---|---|---|
| Edman degradation | N-terminal or C-terminal segment | No | Purified protein or peptide | Direct terminal readout | Limited length; blocked termini reduce success |
| Database-assisted LC-MS/MS | Peptide mapping across known sequence | Yes, reliable reference | Purified protein, construct-matched sample | Efficient confirmation when reference is valid | Weak when sequence differs from database |
| De novo LC-MS/MS | Regional to protein-level assembly | No prior match required | Enriched band, purified target, antibody chain | Recovers sequence without trusted reference | Needs strong spectra and expert review |
| Full-length MS assembly | Broad backbone coverage | Optional; often hybrid | Purified or enriched protein with enough input | Supports near full-length sequence claims | Requires overlap design, depth, and gap reporting |
Hybrid workflows are common in real projects. A team may use database-assisted mapping for most peptides, then apply de novo interpretation to unmatched spectra. Terminal sequencing may confirm the N-terminus while MS mapping covers internal regions. The comparison should therefore include workflow design, not only single-method labels.
Edman Sequencing in Practice
Edman sequencing removes one N-terminal amino acid per cycle and identifies each released residue. The approach is direct and useful when only terminal confirmation is required, such as release testing or N-terminal QC of purified biologics. It is less suited to long internal or full-length recovery because signal loss accumulates and blocked termini can limit success. C-terminal sequencing is a separate specialized workflow and is not interchangeable with N-terminal Edman analysis.
Database-Assisted LC-MS/MS in Practice
A protein is digested, separated by LC, and analyzed by tandem MS. Peptide-spectrum matches are made against a database or supplied construct sequence. When the reference is correct and spectral quality is strong, this path efficiently supports sequence confirmation and coverage mapping for recombinant QC and well-annotated samples. Its main weakness is reference dependence: missing isoforms, variants, or absent entries can prevent valid spectra from receiving confident matches even when MS data are strong.
De Novo LC-MS/MS Sequencing in Practice
De novo sequencing interprets peptide fragmentation without a prior database match. Analysts derive sequence tags from fragment ions, confirm peptides manually, and assemble overlaps from one or more digests. This route fits unknown proteins, antibody variable regions, and samples with incomplete genetic records. It works best on enriched targets with high-quality spectra and is usually applied selectively to unmatched or sequence-critical regions rather than to every spectrum in a complex lysate.
Full-Length MS Assembly in Practice
Full-length protein sequencing requires broad peptide coverage across the backbone. Multiple proteases, repeat MS runs, overlap assembly, and optional terminal confirmation improve confidence. Intact mass and terminal reads can further document truncations or unexpected termini. This path fits projects that need a sequence map rather than a single confirmed peptide, but gap regions and ambiguous residues must be reported explicitly.
Which Strategy Fits Different Research Goals
The best choice depends on what the study must prove.
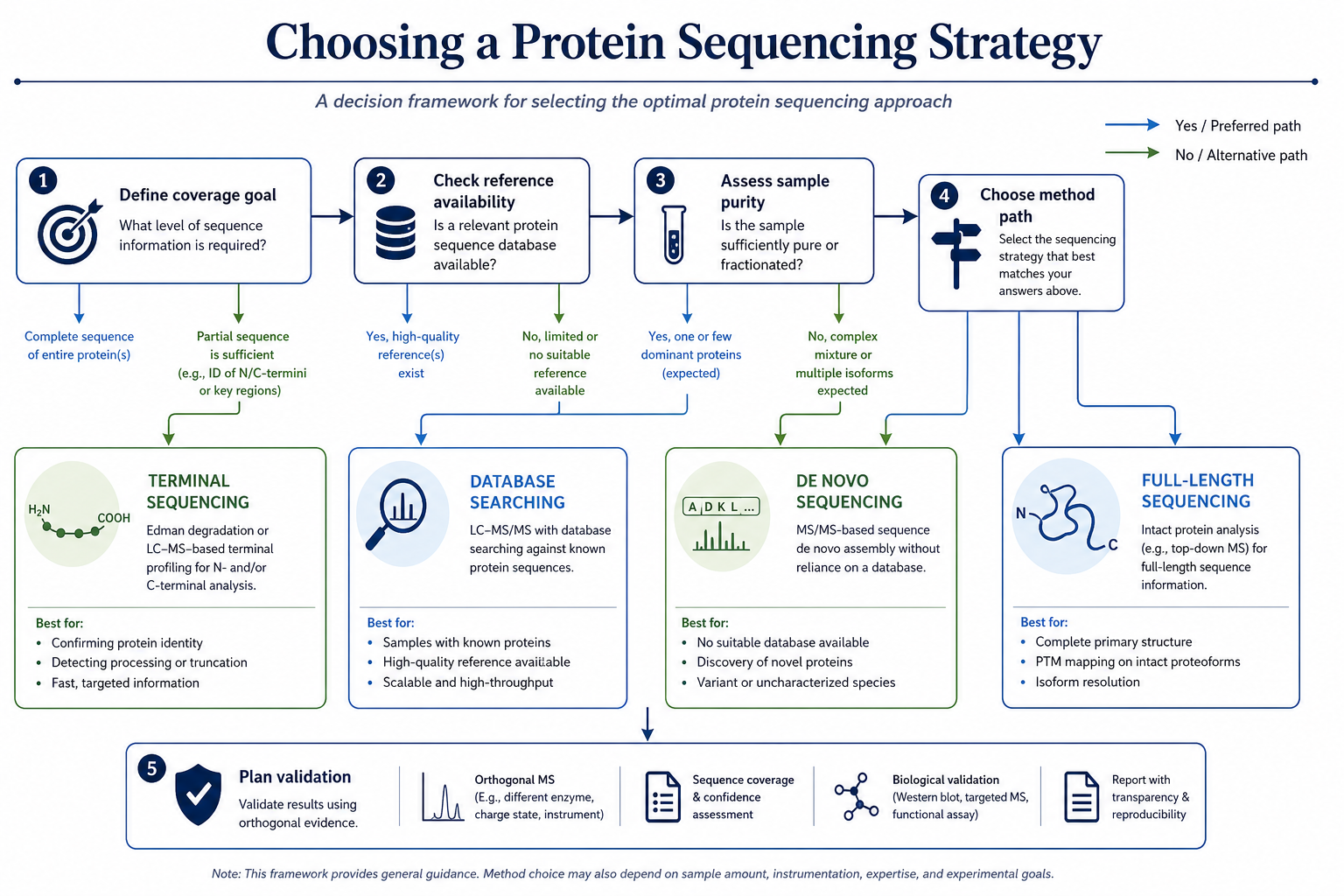
Figure 2. Decision flow for choosing the right sequencing strategy
Reporting depth should be defined before method selection. A terminal result may satisfy one QC gate. A peptide map may satisfy construct confirmation. A de novo or full-length report may be required when the project must support cloning, publication, comparability, or regulatory review.
Research Goal and Method Fit
| Usually Best Starting Point | When to Add a Second Method | |
|---|---|---|
| N-terminal or C-terminal confirmation | Edman sequencing | Add MS mapping if internal coverage is also needed |
| Verify expressed protein matches construct | Database-assisted MS mapping | Add de novo analysis if unmatched variant peptides appear |
| Recover sequence from unknown protein | De novo MS sequencing | Add full-length assembly for broader coverage |
| Document antibody variable region | De novo MS on separated chains | Add terminal sequencing if available |
| Support near full-length sequence claim | Full-length MS assembly | Add Edman or intact mass for termini checks |
| Compare biosimilar or variant-containing product | Hybrid database plus de novo workflow |
Hybrid designs often provide the best balance when most of the sample can be interpreted with a reference but a subset of spectra still requires reference-free analysis.
Limitations and Tradeoffs to Keep in Mind
No single approach is universally superior. Edman sequencing is limited by read length and terminus accessibility. Database-assisted workflows depend on reference quality. De novo sequencing depends on spectrum quality and expert review. Full-length assembly depends on overlap coverage and transparent gap reporting. Identification coverage and sequence proof are not the same deliverable.
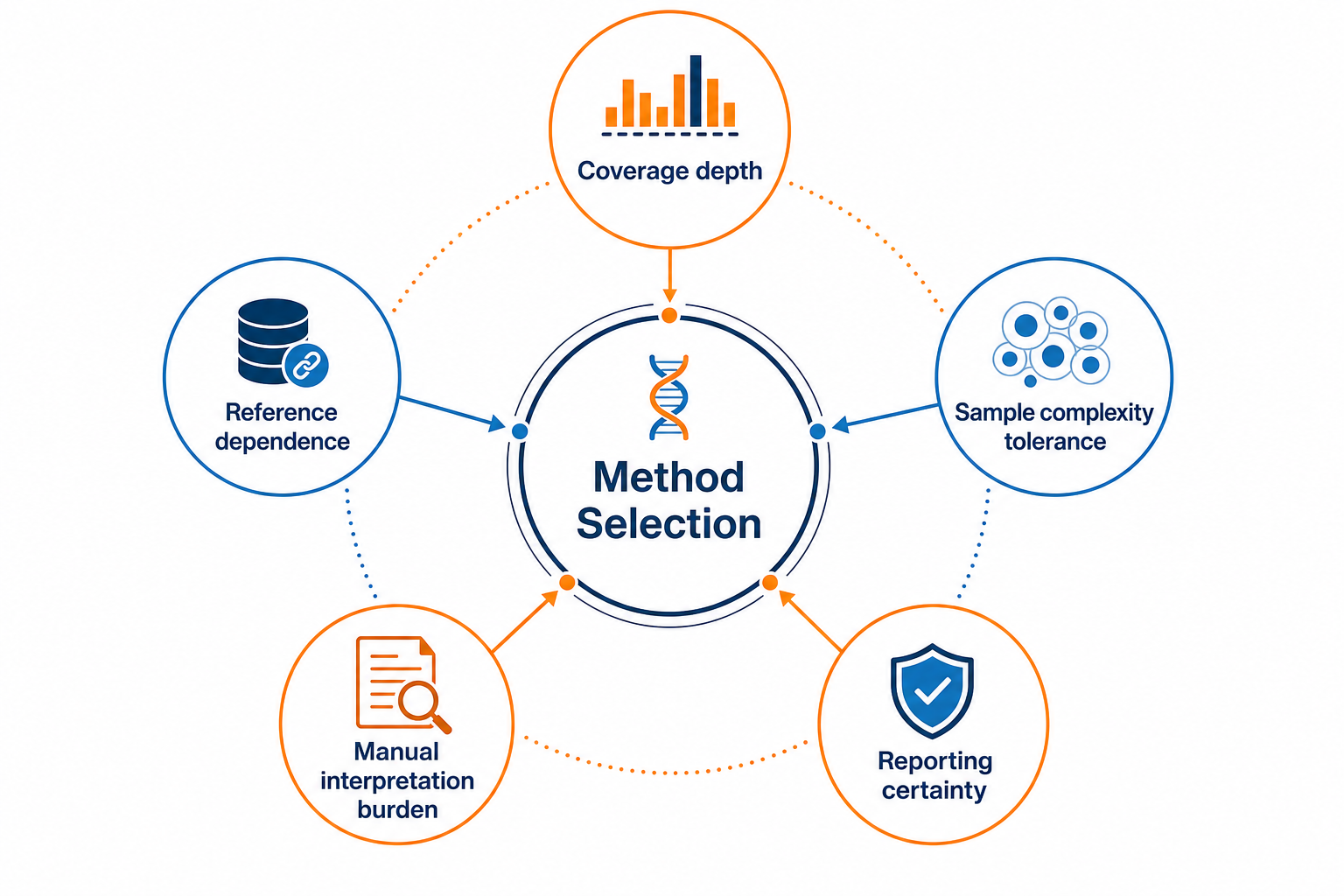
Figure 3. Key tradeoffs to weigh when selecting a sequencing strategy
Before committing sample and MS time, confirm whether the required evidence is terminal, regional, or full-length; whether a reliable reference exists; whether the sample is purified or complex; and whether the report must support QC, cloning, or regulatory use. Matching protein sequencing methods to these constraints is usually more reliable than defaulting to the most familiar lab workflow.
Frequently Asked Questions
1. What are the main protein sequencing methods?
The most common approaches are Edman sequencing for terminal reads, database-assisted LC-MS/MS for reference-based confirmation, de novo LC-MS/MS for reference-free interpretation, and full-length MS assembly for broader backbone coverage.
2. Is Edman sequencing still useful?
Yes. Edman sequencing remains valuable for terminal confirmation when the sample is purified and only end-sequence evidence is required. It is less suited to full-length internal sequencing.
3. When should I choose de novo sequencing over database search?
Choose de novo sequencing when the protein sequence may be absent, incorrect, proprietary, or engineered in a way that makes database matching unreliable.
4. Do I need full-length sequencing for every project?
No. Many projects need only terminal confirmation or regional peptide coverage. Full-length assembly is most appropriate when the deliverable is a broader sequence map with overlap support.
5. Can one project combine multiple methods?
Yes. Hybrid workflows are common. Terminal sequencing, database mapping, and de novo interpretation are often combined when different regions of the same protein require different evidence standards.
Conclusion
Protein sequencing methods serve different research needs. Edman sequencing fits terminal confirmation. Database-assisted tandem MS fits construct verification when references are reliable. De novo sequencing fits unknown, engineered, or variant-containing proteins. Full-length assembly fits projects that require broader sequence documentation with overlap support.
If your project sits at the boundary between identification and sequence proof, contact MtoZ Biolabs to discuss whether terminal sequencing, database-assisted mapping, de novo protein sequencing, full-length assembly, or a combined MS workflow is the right fit.
How to order?

