





PhIP-Seq Data Analysis Workflow: From Read Counts to Enriched Peptide Hits
- the peptide shows enrichment above the negative control distribution after normalization
- recurrent signal in bead-only control or mock IP has been filtered, flagged, or down-ranked
- replicate concordance is acceptable for the study design
- the peptide remains interpretable in context of neighboring overlapping peptides, count sparsity, and library representation
- sample and control identifiers, including bead-only control or mock IP when used
- key QC metrics such as mapped reads, retained reads, and count sparsity
- normalization method and any reference library comparison
- background filtering logic for nonspecific binders
- enrichment scoring definition and threshold rationale
- replicate concordance review
- ranked enriched peptide hits with region-level grouping of overlapping peptides
- explicit notes on interpretation limits and validation planning
A practical phip seq data analysis workflow starts with count QC review, then moves through normalization, background filtering, enrichment scoring, replicate concordance, and hit ranking. That order matters because PhIP-Seq measures relative enrichment of peptide-displaying phage after immunoprecipitation, not direct protein abundance. If teams sort peptides from raw read counts too early, library bias and recurrent background binders can take over the final list.
The post-sequencing problem is simple to state but harder to handle consistently. A team may already have FASTQ files or a peptide count matrix from immunoprecipitated samples, reference material, and negative controls, yet still have no clear rule for deciding which peptides reflect meaningful antibody binding. The aim is not the longest hit list. It is a ranked set of enriched peptide hits that separates from control behavior, holds up across replicates, and is suitable for validation planning.
Where PhIP-Seq Interpretation Often Breaks Down
Many groups already know how to map reads and summarize counts. The harder part is deciding what those counts mean in a phage-displayed peptide library. One sample may have high depth but still be weak at the peptide level because most reads collapse into a few dominant clones. Another may produce many apparent hits that also recur in the bead-only control. Replicates can match on total mapped reads yet disagree on which peptides look enriched.
That is why PhIP-Seq should not be treated like a standard RNA-seq comparison. The evidence unit is the peptide, often strengthened by overlapping peptides that point to a linear epitope region. A convincing result is usually not one isolated spike. It is a pattern that stays separated from the negative control, survives background filtering, and shows acceptable replicate concordance.
The Main Causes of Weak or Misleading Hit Calls
Uneven library representation
A phage-displayed peptide library rarely starts with uniform abundance. Some peptides are overrepresented before immunoprecipitation, while others are barely observed. Raw read counts therefore mix starting library composition with sample-specific enrichment.
Recurrent nonspecific binders
Certain clones repeatedly score in a negative control, bead-only control, or mock IP. If those peptides are not modeled directly, they can rise in the rankings even when the signal is not sample specific.
Replicate instability
Borderline peptides often appear in one replicate and disappear in another. That pattern may come from stochastic sampling, sparse counts, or unstable immunoprecipitation performance rather than durable antibody binding.
Threshold logic that ignores peptide context
A single cutoff applied after depth scaling can miss the structure of PhIP-Seq data. Useful hit calling also needs background separation, count sparsity, and support from overlapping peptides.
Related Services
Main Service |
Supporting Service |
Validation Service |
Alternative Service |
Step-by-Step Workflow for Turning Counts Into Hits
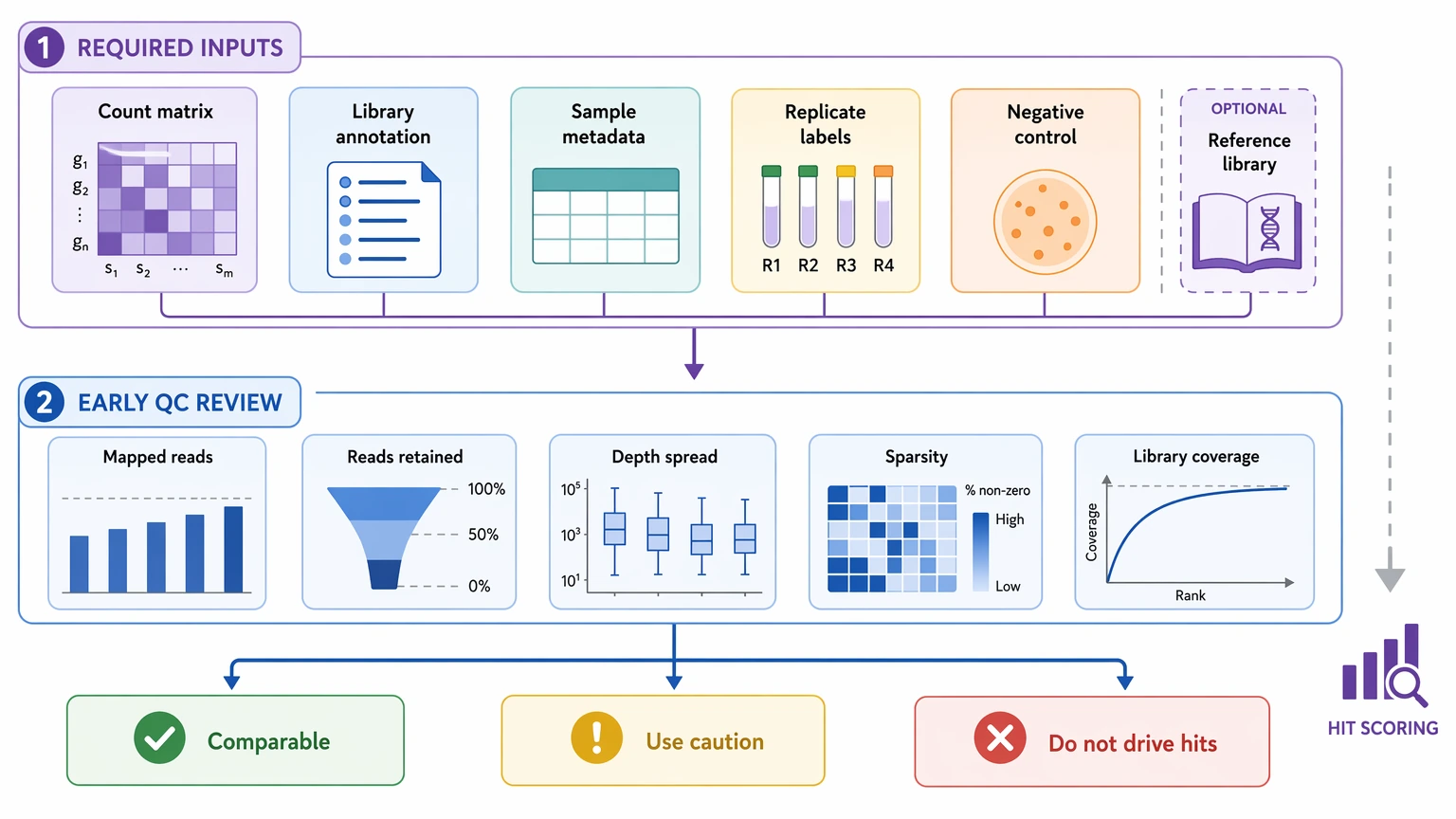
1. Confirm the minimum analysis inputs
Before scoring anything, verify that the dataset includes mapped read counts or a peptide count matrix, peptide library annotation, sample metadata, and replicate labels. A negative control is also important, ideally a bead-only control or mock IP. If input or reference library counts are available, keep them in the same analysis set rather than moving them into a separate appendix.
At this stage, review total mapped reads assigned to library peptides, reads retained after preprocessing and mapping, sample-to-sample depth spread, peptide-level sparsity, and broad library representation. The point is not to pass or fail a sample on one metric. It is to separate samples that are reasonably comparable from those that need caution, and from those that should not drive hit calling.
2. Define what counts as a reportable signal
The next step is conceptual: what will the final report call a hit? In PhIP-Seq, the evidence unit is an enriched peptide hit, but many projects benefit from a second layer of region-level interpretation. If several overlapping peptides from the same antigen segment enrich together, that pattern usually carries more weight than one isolated peptide with no local support.
Definition of a reportable enriched peptide hit:
That definition should match the project stage. An exploratory discovery screen may keep a wider ranked set for review. A validation-oriented shortlist should be tighter and put more weight on negative-control separation, replicate concordance, and contiguous overlapping peptides. If your team needs an external review of scoring logic or reporting criteria, submit your requirements to MtoZ Biolabs with the peptide count matrix, control structure, replicate layout, and intended follow-up assay so the analysis plan can be matched to the study.
3. Normalize with library behavior in mind
Normalization should reduce sequencing depth differences, but it also has to respect the fact that a peptide library is not an evenly distributed transcriptome. Simple count scaling can help, yet it should be followed by distribution review at the peptide level. Otherwise, dominant library members may still shape the ranking after normalization.
When reference library data exist, compare immunoprecipitation samples against that baseline to separate enrichment from starting abundance. Also flag peptides that stay consistently absent, extremely low count, or disproportionately dominant across unrelated samples. Those features do not automatically disqualify a peptide, but they do change how much confidence its score deserves.
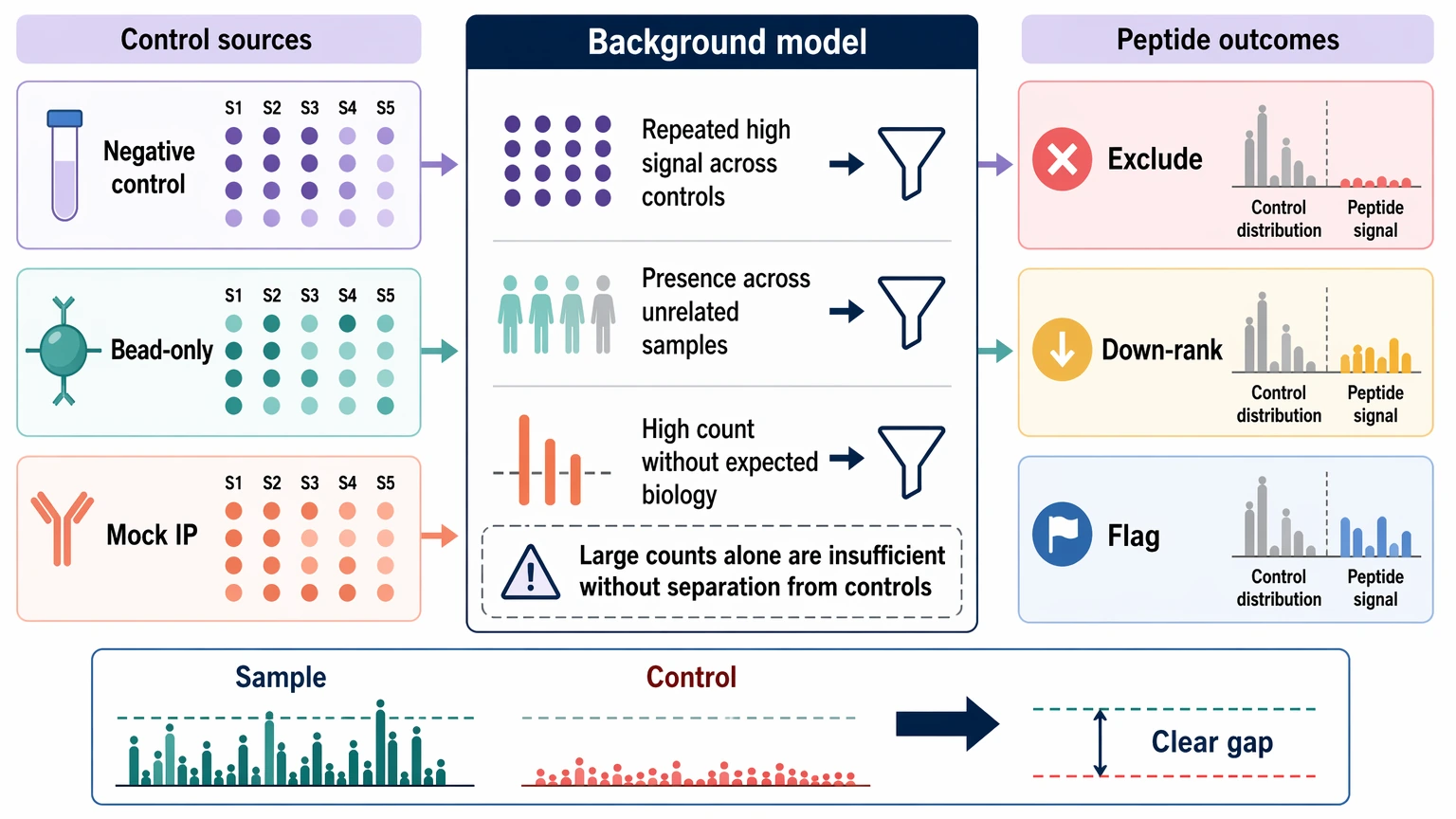
4. Build background filtering from observed controls
Background filtering is the most PhIP-Seq-specific step. Use the negative control, bead-only control, and mock IP to model nonspecific binders rather than treating them as a last sanity check. Analysts should look for peptides that repeatedly enrich across controls, appear across many unrelated samples, or stay high even when biological binding is not expected.
Those recurrent background peptides can then be excluded, down-ranked, or flagged in the output table. The exact rule depends on study design and library structure, but the core idea does not change: a peptide should not be promoted just because it has a large count. It should show a visible gap between experimental samples and the control distribution.
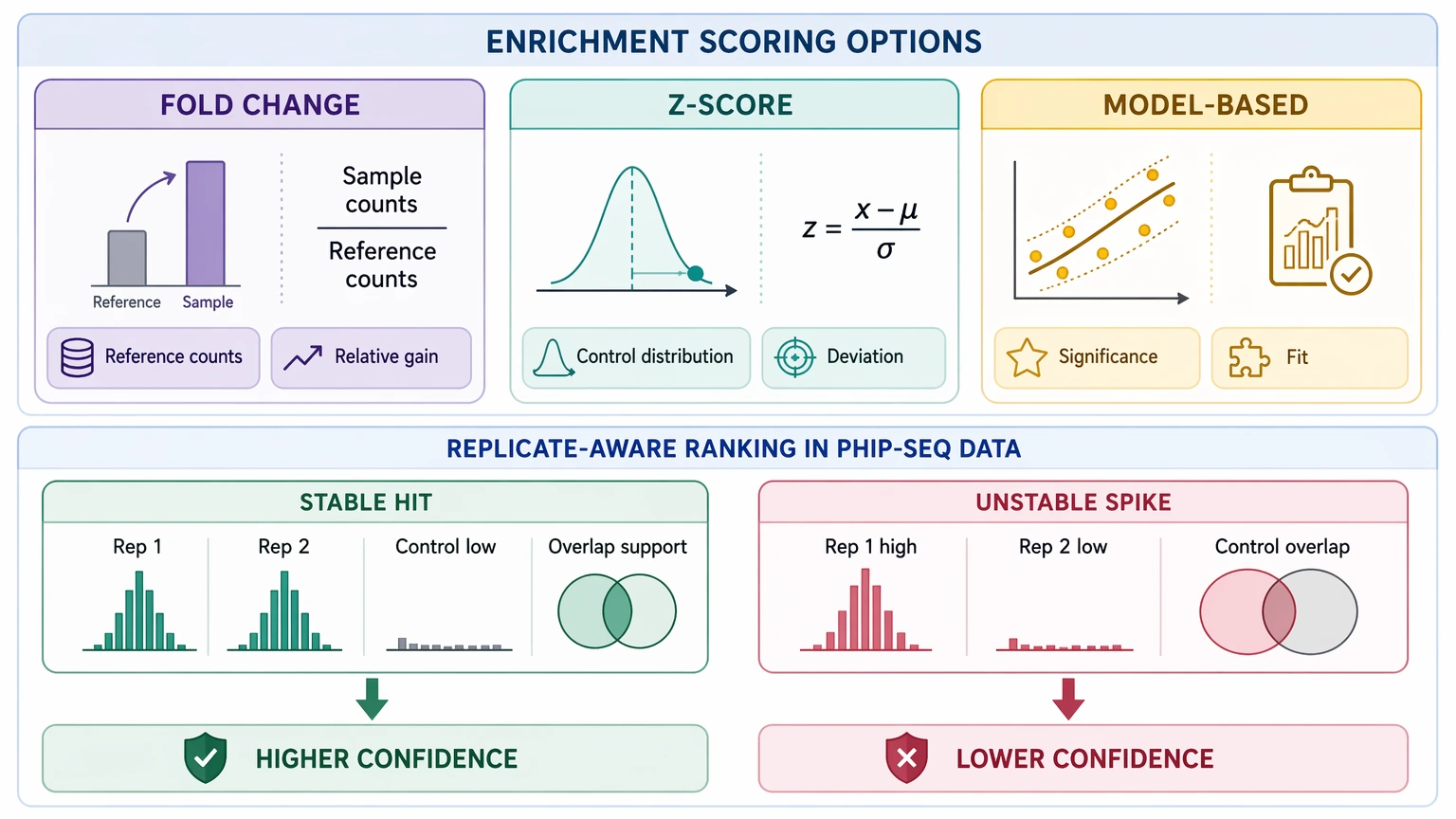
5. Apply enrichment scoring with replicate awareness
After normalization and background filtering, calculate enrichment scoring with a method that fits the dataset. Some projects use fold-change-like measures against reference counts, some use z-score-style comparison against control distributions, and others use model-based significance scoring. No single framework fits every study, so transparent reporting matters more than forcing one metric onto every dataset.
Replicate concordance should shape final confidence. A peptide observed across replicates, separated from the negative control, and supported by nearby overlapping peptides should rank above a larger but unstable single-replicate spike. Before locking thresholds, review how many peptides survive per sample, how often control-associated peptides still sit near the top, and whether the retained list remains interpretable at the peptide level.
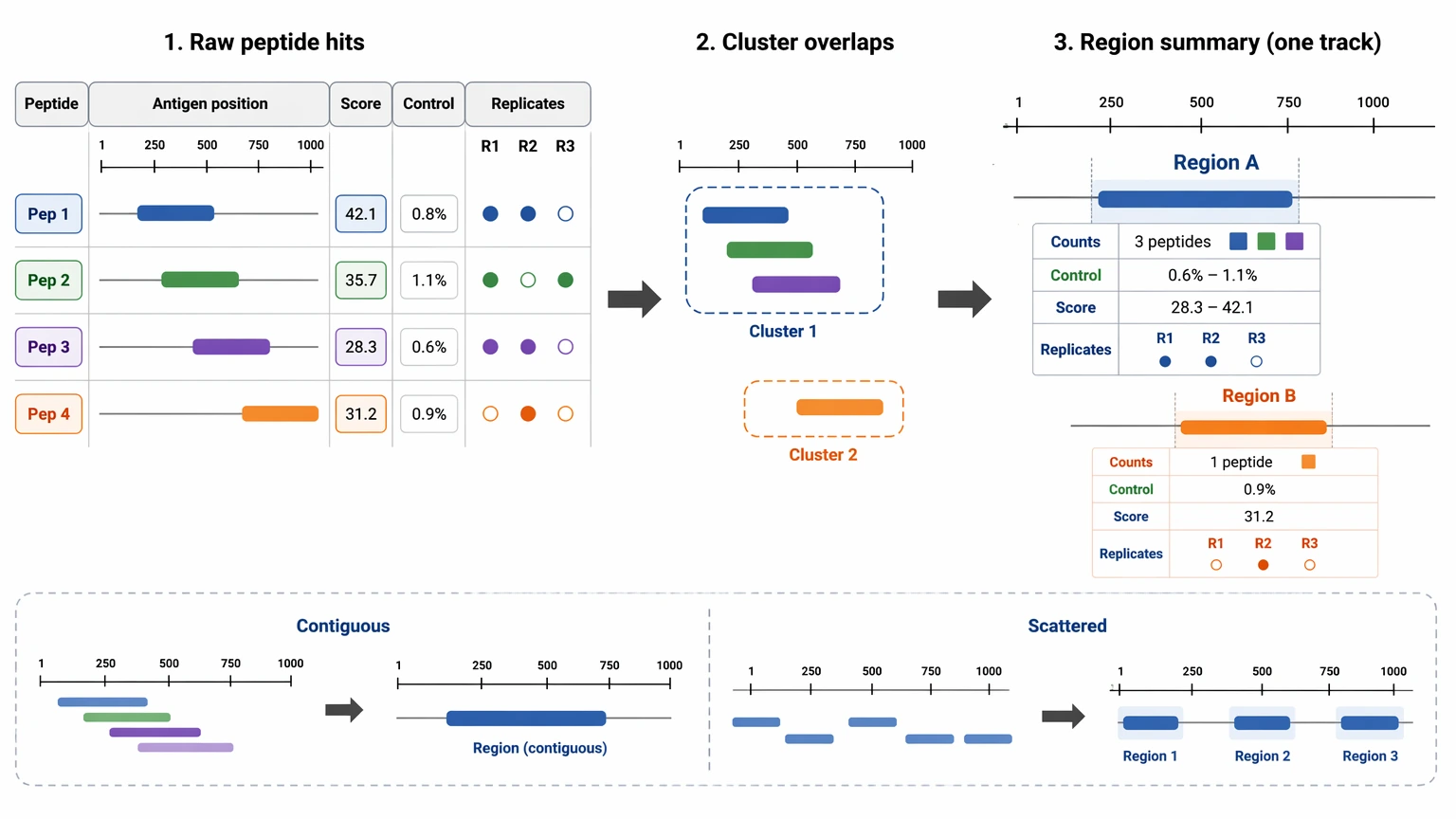
6. Collapse redundant peptide hits into region-level summaries
A raw hit table can overstate the number of findings when multiple overlapping peptides represent the same linear epitope region. Instead of listing each peptide as a separate discovery, cluster overlapping peptides and summarize region-level support. That makes the output easier to review and better aligned with how validation planning usually moves forward.
Region-level summaries should still preserve peptide-level evidence. A useful report shows the underlying peptide count matrix values, control behavior, enrichment scoring, and replicate concordance for each member of the cluster. That lets reviewers judge whether the region reflects a contiguous pattern or a scattered set of weak events.
What a Defensible Output Report Should Include
A useful final report does more than rank peptides by score. It should document preprocessing retention, mapped read assignment, normalization logic, control-derived background filtering, scoring method, and replicate handling. It should also identify peptides removed as nonspecific binders, peptides retained with caution, and clusters of overlapping peptides that support linear epitope interpretation.
Minimum elements of a defensible output report:
For internal decisions, the main question is whether the top hits still look defensible after each filter layer. If a peptide disappears once controls are considered, or if its score depends on a single replicate, it may still be worth noting but not prioritizing for immediate follow-up. By contrast, repeated enrichment with adjacent peptide support is a better starting point for validation planning.
Interpretation Boundaries to Keep in Mind
Even a careful workflow cannot rescue weak wet-lab design or poor control selection. Underrepresented peptides, unstable immunoprecipitation, or missing negative control data can all limit confidence in the final ranking. The analysis should make those limits visible rather than hiding them behind one summary score.
It is also important to keep the biological claim narrow. PhIP-Seq supports peptide-level interpretation and can help localize a linear epitope, but it does not directly measure absolute antibody concentration and does not comprehensively capture conformational epitope binding. When the strongest claim rests on one peptide or an unexpected antigen region, orthogonal confirmation is the sensible next step, not an optional extra.
Conclusion
A reliable PhIP-Seq workflow moves from QC and input review to normalization, control-based background filtering, replicate-aware enrichment scoring, and region-level hit ranking. This structure fits antibody profiling projects that already have sequencing output but still need a defensible shortlist for peptide-level interpretation and validation planning. For studies where ranked hits will guide follow-up assays, submit your requirements to MtoZ Biolabs and include the count matrix format, control types, replicate structure, library annotation, and downstream validation goal so the team can evaluate your project and discuss an analysis and reporting approach that matches the data's strengths and limits.
FAQ
Can one negative control support an entire multi-batch PhIP-Seq study?
Usually not by itself. Background structure can shift across batches, bead lots, or immunoprecipitation runs. A control set that reflects the actual processing batch gives a more trustworthy baseline for background filtering.
When should isolated single-peptide hits be kept on the ranked list?
Keep them when they remain clearly separated from controls, show acceptable replicate concordance, and fit the library design, but label them as lower-confidence candidates than contiguous overlapping peptide patterns. They often work better as follow-up questions than headline findings.
How do underrepresented library members affect downstream interpretation?
A poorly represented peptide can look absent even when antibodies are present, or appear unstable across replicates because sampling noise is high. That is why library representation should be reviewed before interpreting non-enrichment as a biological negative.
Should sample ranking be based on the number of enriched peptides?
Not by count alone. A sample with fewer hits may still be more convincing if those hits are reproducible, well separated from controls, and clustered across overlapping peptides. Hit quality matters more than list length.
What information should we prepare before asking for outside analysis support?
Prepare the peptide count matrix or mapped read table, sample sheet, replicate labels, library annotation, details on reference material, and all negative control data. It also helps to state whether the next decision is exploratory ranking, antigen-region review, or orthogonal validation planning.
How to order?

