





PhIP-Seq Analysis: Normalization, Background Control, and Hit Calling Decisions
Start with the negative-control model, then choose read count normalization that can handle sparse peptide counts, and only after that apply combined hit calling criteria. In PhIP-Seq analysis, that sequence is usually the quickest way to tell whether peptide-level enrichment points to plausible antibody binding or to a mix of library bias, background reactivity, and low-count noise.
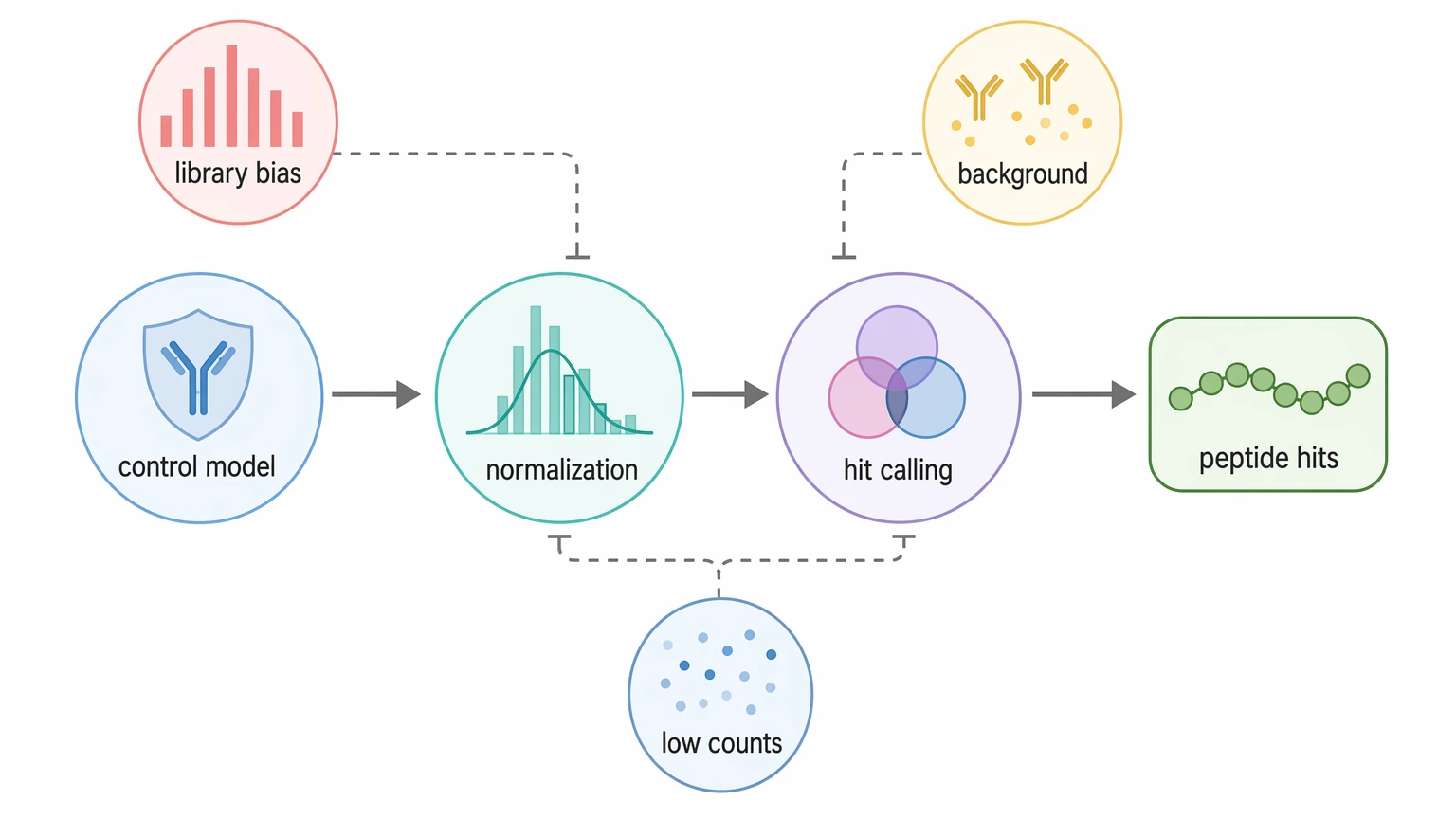
That order matters because PhIP-Seq does not behave like routine RNA-seq. The readout comes from a phage-displayed peptide library after immunoprecipitation and next-generation sequencing, so apparent signal can shift fast when the baseline, scaling rule, or threshold changes. For translational teams, the goal is not the biggest statistic on the page. It is a hit list that still looks defensible when you line up replicates, controls, and follow-up validation.
Where PhIP-Seq Interpretation Usually Breaks Down
Most trouble shows up after sequencing, when the dataset appears usable but the enrichment pattern will not settle down. One replicate may rank a peptide near the top, while another keeps it close to background. A mock IP or bead-only control may also carry broad signal across peptides that were expected to stay quiet. At that point, analysts are left asking whether the assay captured specific antibody binding or just repeated non-specific pull-down.
Once that uncertainty appears, it affects every downstream choice. Total-read scaling can favor already abundant clones. Fold-enrichment can blow up when the starting count is near zero. A z-score-like statistic may look persuasive until the same peptide appears on and off across negative controls. If those issues stay unresolved, protein-level summaries and linear epitope claims can end up sounding firmer than the data really are.
The Main Causes of Unstable Results
Four categories explain most inconsistent PhIP-Seq outputs in this setting.
First, library bias shifts the baseline before enrichment is even measured. In a phage-displayed peptide library, some clones are more abundant at input and some propagate more efficiently. If baseline representation is uneven, simple depth scaling can overstate peptides that were already common and downplay peptides that started low but enriched consistently.
Second, background reactivity depends on the control design. A bead-only control captures stickiness to beads and reagents. Mock IP captures a broader layer of immunoprecipitation-related background. A biological negative control reflects sample-matrix effects and low-level reactivity that buffer-only controls cannot show. Those controls are not interchangeable because they describe different processes.
Third, sparse counts make ratios unstable. Many peptides sit at zero or near-zero counts, so even a small absolute increase can create a large fold-enrichment value. Without low-count filtering and variance-aware scoring, those features can crowd the hit list even when the biological case is weak.
Fourth, one-dimensional hit calling raises false-positive risk. Fold-enrichment alone, FDR alone, or a z-score-like statistic alone cannot fully handle sticky peptides, peptide dropout, or weak replicate concordance. In PhIP-Seq, confidence comes from agreement across several checks, not from one universal cutoff.
Related Services
Main Service |
Supporting Service |
Validation Service |
Alternative Service |
A Stepwise Framework for Data Interpretation
1. Inspect raw count structure before modeling enrichment
Start with total sequencing reads per sample and per negative control, the fraction of zero-count peptides, and the spread of counts across the library. That first pass tells you whether depth imbalance, severe sparsity, or a few highly represented clones are dominating the dataset.
If pre-immunoprecipitation library data are available, compare them with post-immunoprecipitation counts. That helps separate depth effects from true library bias and can flag peptide dropout that will distort later statistics. One useful checkpoint is whether the same small subset of peptides absorbs a large share of reads across many samples. If that pattern repeats, downstream normalization has to account for clone representation directly rather than assume a balanced starting pool.
2. Define what each negative control is measuring
Before background correction, state what question each control is answering. A bead-only control estimates non-specific binding to beads and reagents. Mock IP captures broader workflow background from the immunoprecipitation setup. Biological negative control samples estimate matrix-linked background reactivity that the other two controls cannot show.
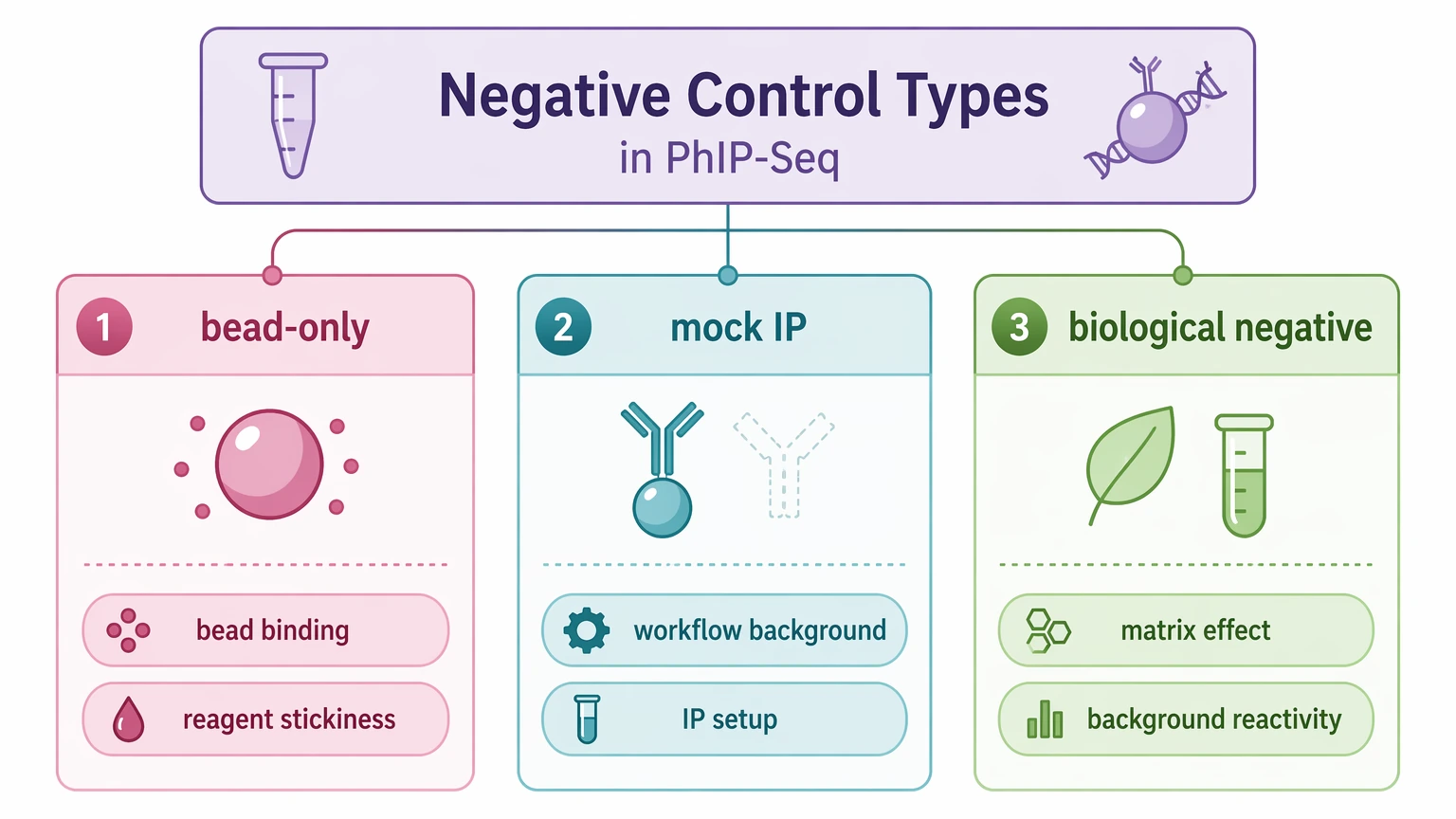
Published PhIP-Seq workflows often keep these control types separate because each one anchors a different empirical null. If the same peptides recur across bead-only or mock IP samples, those peptides often behave more like recurring assay background than new antibody targets. If that decision is blocking progress, contact MtoZ Biolabs to submit your requirements and review whether the current control set matches the project objective and count structure.
3. Choose read count normalization that fits sparse peptide data
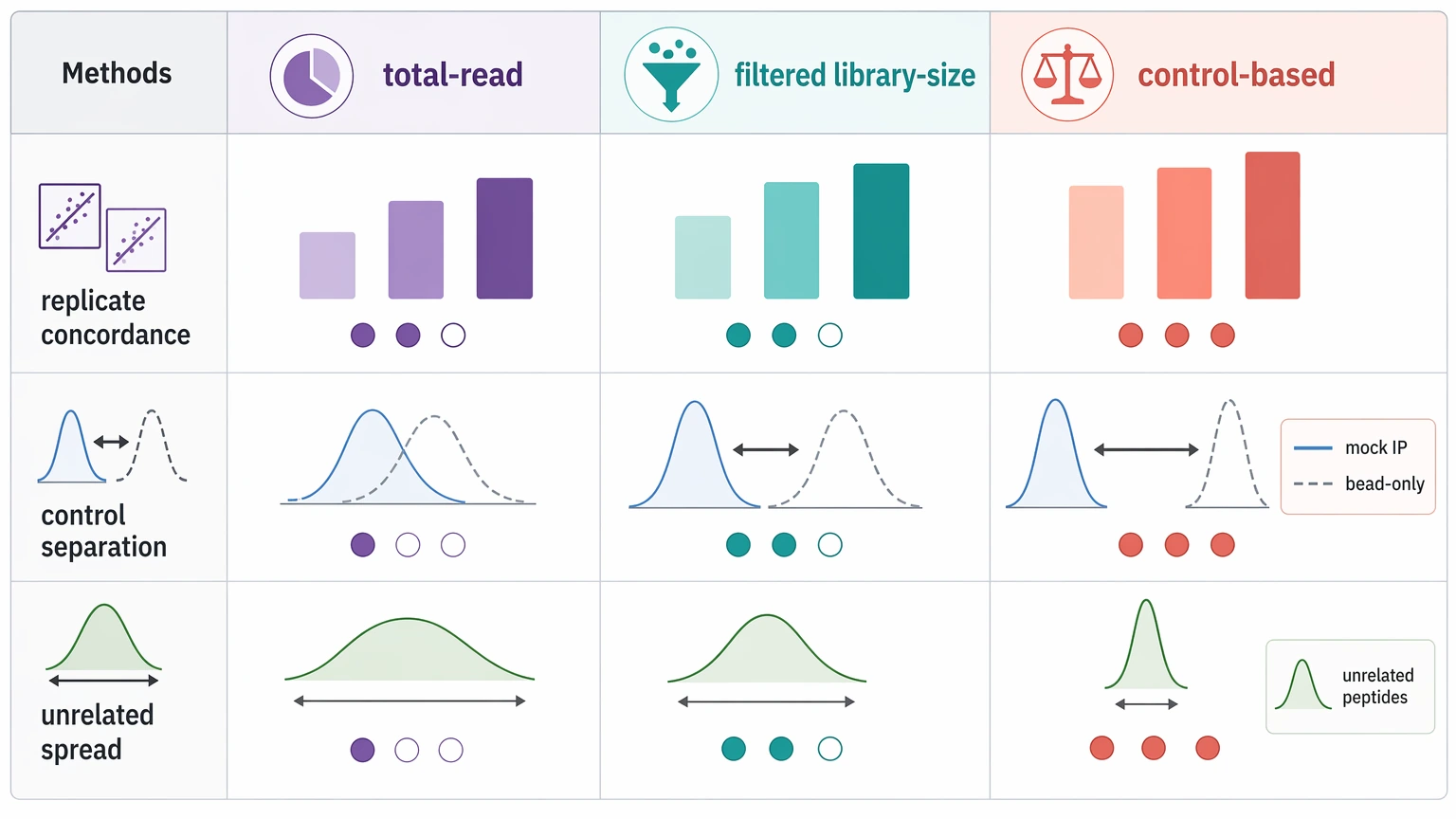
Read count normalization in PhIP-Seq is not just a matter of equalizing library size. It also has to keep low-count features from becoming artificially dominant. Total-read scaling is a reasonable place to start, but it is rarely enough by itself.
A better approach is to compare several normalization strategies side by side: total-read scaling, library-size adjustment after low-count filtering, and control-based scaling anchored to mock IP or bead-only control distributions. The best option is the one that improves replicate concordance and sharpens separation from the selected negative control without creating broad enrichment across unrelated peptides. Reanalysis frameworks for PhIP-Seq often favor this direct comparison instead of assuming one method will work across all libraries.
4. Score peptide-level enrichment against the empirical null
After normalization, evaluate peptide-level enrichment against the selected empirical null. This may involve fold-enrichment, a z score or z-score-like statistic, or another model that uses control variance. The exact score matters less than whether it behaves sensibly across controls and replicates.
A large statistic by itself is not enough. Check whether the peptide stays separated from the negative control distribution and whether the signal persists across technical or biological replicates. A one-time spike with poor replicate concordance is weak evidence for specific antibody binding, especially when that peptide shows up intermittently in background controls. By contrast, moderate enrichment that repeats cleanly is often a better follow-up candidate.
5. Perform hit calling with combined filters
Reliable hit calling usually combines low-count filtering, enrichment relative to the most relevant negative control, acceptable false discovery rate (FDR) behavior, and replicate concordance. Some projects also require support from more than one peptide within the same antigen region or epitope cluster before moving from a peptide-level observation to an antigen-fragment interpretation.
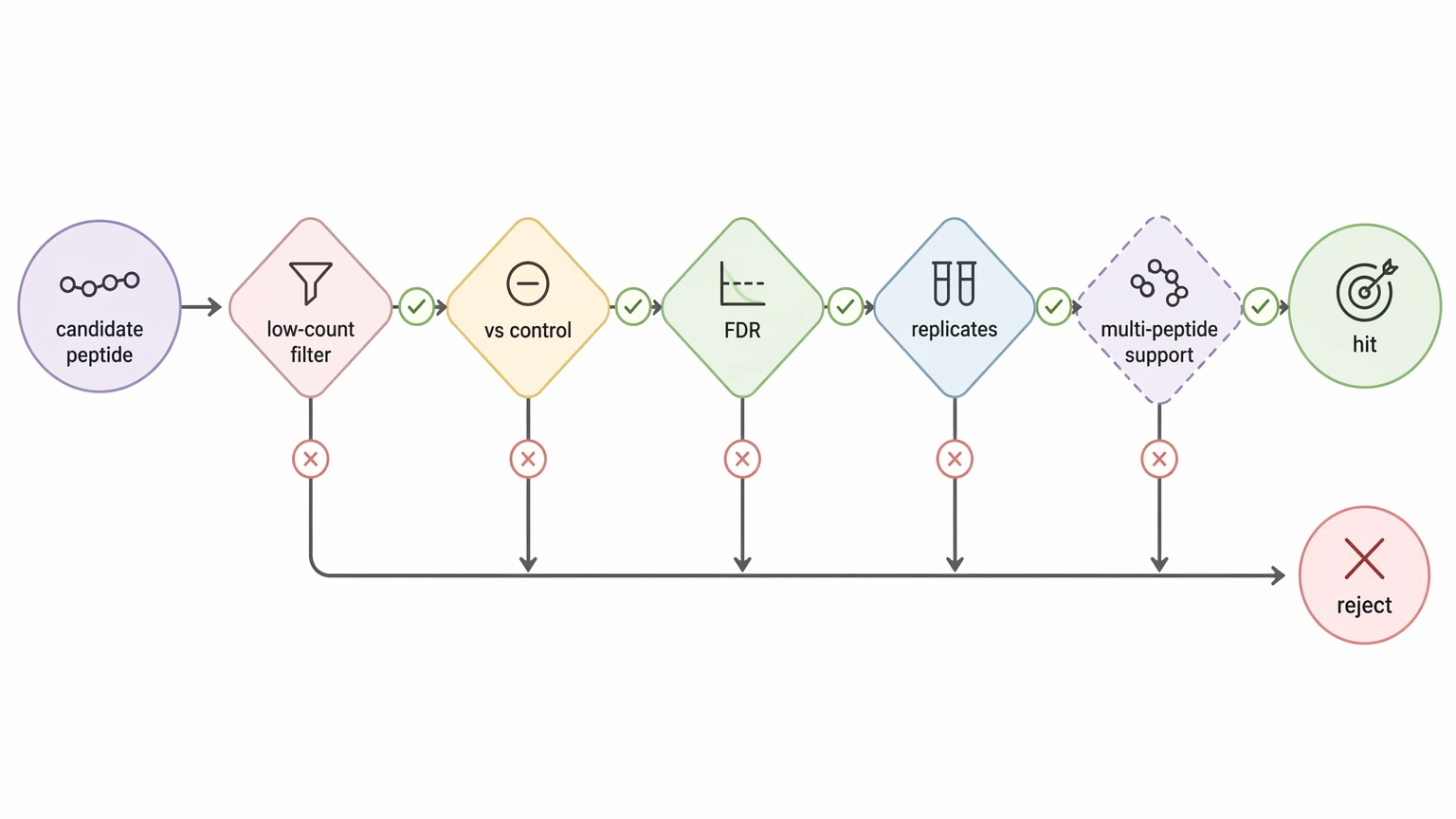
This combined logic cuts down false positives from sticky peptides and clone-specific artifacts. It also yields a hit list that is easier to defend in translational work, where peptide calls shape what gets validated next. If a small threshold change completely rewrites the hit list, the analysis still needs work before strong biological claims are made.
6. Separate statistical hits from validation-ready candidates
The last step is triage. PhIP-Seq measures relative peptide-level enrichment associated with linear epitope recognition. It does not provide calibrated antibody concentration, and it does not fully capture native conformational binding.
Candidates are usually stronger when they satisfy three conditions: they stay separated from background reactivity under the chosen control model, they show replicate concordance, and they fit a coherent linear epitope or epitope cluster pattern. Reviews of peptide-display serology also keep making the same point: enriched peptides should be treated as screening outputs until orthogonal validation confirms the biological interpretation.
What a Successful Reanalysis Should Look Like
A useful reanalysis should produce visible changes, not just a new spreadsheet. Negative control distributions should separate more clearly from test samples. Extreme scores driven by near-zero denominators should become less common. Peptide rankings should also become more stable across replicates and under small parameter changes.
Check both numerical and biological signals. Numerically, you want better replicate concordance, fewer recurring hits shared with mock IP or bead-only control samples, and more stable significance patterns after low-count filtering. Biologically, you want enriched peptides to overlap within a plausible antigen region rather than scatter randomly across unrelated proteins.
Practical Boundaries Before Moving Forward
Some datasets will not become interpretable through reprocessing alone. Very low immunoprecipitation yield, severe peptide dropout, or highly uneven sequencing depth can leave too little information to define a stable empirical null. In that situation, adding controls or redesigning part of the experiment may be more productive than tightening thresholds around noisy data.
Batch context matters too. A recurring peptide pattern may track with carryover, reagent background, or batch-specific library behavior rather than cohort biology. PhIP-Seq analysis is most useful when normalization, background control, and hit calling are treated as one linked decision system. That framing fits projects deciding whether current peptide-level enrichment is strong enough for linear epitope review or orthogonal validation. If your dataset is at that stage, contact MtoZ Biolabs to submit your requirements and discuss the control strategy, replicate structure, and follow-up validation plan.
Conclusion
The most defensible PhIP-Seq hit lists come from a fixed interpretation order: define the empirical null, normalize with sparse counts in mind, and then call hits with combined filters rather than one statistic alone. That approach is most useful for antibody profiling projects where control separation, replicate behavior, and peptide-level consistency matter more than headline scores. For studies at the reanalysis or validation-planning stage, submit your requirements to discuss sample context, control design, and the next validation step before moving peptide candidates into stronger biological claims.
FAQ
Should technical replicates and biological replicates be treated the same way?
No. Technical replicates test assay and sequencing consistency, while biological replicates reflect real sample variation. A peptide that fails technical replicate concordance is usually a weak analytical hit.
When does low-count filtering become too aggressive?
It becomes too aggressive when reproducible peptides are removed only because they begin from a low-abundance library state. The goal is to suppress unstable denominator effects, not erase sparse but repeated enrichment.
Can protein-level summaries replace peptide-level hit calling?
No. Protein-level summaries help organize results, but they should not rescue weak peptide evidence. The primary unit of evidence remains the peptide-level enrichment pattern against controls and replicates.
What is a warning sign that the empirical null is poorly defined?
Warning signs include broad variance across negative controls, frequent apparent enrichment inside the control set, or score behavior that changes sharply between batches. Those patterns suggest the background model is missing the main noise source.
When should an alternative platform be considered?
An alternative platform is worth considering when control separation stays weak, peptide dropout remains heavy, or the biological question depends on conformational rather than linear epitopes. In that setting, a protein array or another orthogonal assay may fit better.
How to order?

