





NGS Sequencing De Novo: When Short-Read Data Are Enough and When You Need a Hybrid Assembly Design
- the project needs full-length transcript models rather than partial CDS fragments
- isoform-specific interpretation changes the conclusion
- the sample contains many related paralogs or repetitive coding regions
- mature peptides must be linked back to precursor architecture
- the target includes rearranged, variable, or engineered sequence segments
- assembled transcripts or contigs
- predicted proteins from CDS and ORF calling
- a ranked candidate protein sequence list
- notes on ORF completeness, isoform ambiguity, or suspected chimeric assembly
- mapping summaries that show where LC-MS/MS peptides support each candidate
- enough full-length transcript recovery for the main candidates
- acceptable sequence coverage across the relevant ORFs
- enough unique peptide mapping to reduce competing protein assignments
- a realistic next validation step rather than another round of uncertain interpretation
Short-read ngs sequencing de novo is often enough when the goal is broad coding sequence (CDS) recovery from a moderately complex transcriptome and the downstream LC-MS/MS question does not hinge on exact full-length transcript structure. A hybrid assembly becomes easier to justify when fragmented contigs, weak isoform resolution, or poor unique peptide mapping would directly weaken protein inference.
Quick Decision Guide
| Project signal | Better starting design | Why it fits |
|---|---|---|
| Moderate transcript complexity, good RNA, main need is candidate CDS recovery | Short-read sequencing | Usually sufficient for broad transcriptome assembly and ORF prediction |
| Novel proteins suspected, but isoform specificity is not central | Short-read sequencing first | Lets the team test whether assembly contiguity is already adequate |
| Paralog-rich families, splice complexity, rearranged regions, or engineered constructs | Hybrid assembly | Improves transcript continuity and helps separate structurally similar candidates |
| Secreted peptide, venom, antibody-like, or PTM-rich targets with ambiguous mapping | Often hybrid assembly | Reduces transcript-level ambiguity before de novo peptide sequencing follow-up |
Use this comparison as a workflow decision, not a platform ranking. The real question is whether short-read sequencing can produce candidate protein sequences that are clear enough for database search rescue, de novo peptide sequencing, and a realistic validation workflow.
What “Enough” Means in a De Novo Protein Discovery Project
For reference-free protein discovery, “enough” is not a large transcript set or a favorable assembly statistic on its own. It means the sequencing design supports the biological claim you want to make.
In practice, short-read sequencing is enough when four conditions mostly hold. First, the transcriptome assembly yields usable open reading frames (ORFs) for the targets that matter. Second, the main candidates do not rely on exact splice-form assignment. Third, peptide evidence maps to a manageable number of predicted proteins. Fourth, the remaining uncertainty still fits the next validation step.
That last point is easy to miss. Even when LC-MS/MS data look convincing, predicted proteins from a de novo assembly are still inferential. Peptide-spectrum matches can support a sequence region, but they do not always prove the exact full transcript structure, especially when post-translational modification (PTM) signals, homologous proteins, or incomplete assemblies complicate interpretation.
Why Short-Read-Only Designs Often Work
Short-read sequencing remains a sensible first move for many reference-free workflow designs. If the transcript population is not highly repetitive or packed with isoforms, paired-end short reads can support solid transcriptome assembly, broad CDS recovery, and a practical protein candidate database.
This is especially useful when the team needs to expand the search space after poor database search coverage. A short-read assembly can add enough CDS and predicted protein content to reinterpret LC-MS/MS data, rank candidate sequences, and decide which signals deserve targeted follow-up.
Short-read sequencing also works well in staged planning. Teams can assemble transcripts, check assembly contiguity, review ORF completeness, and add long reads only if protein-relevant weaknesses remain. That is often a reasonable path when budget or RNA amount is limited and the open question is whether the transcript side is usable, not whether every structure is fully resolved.
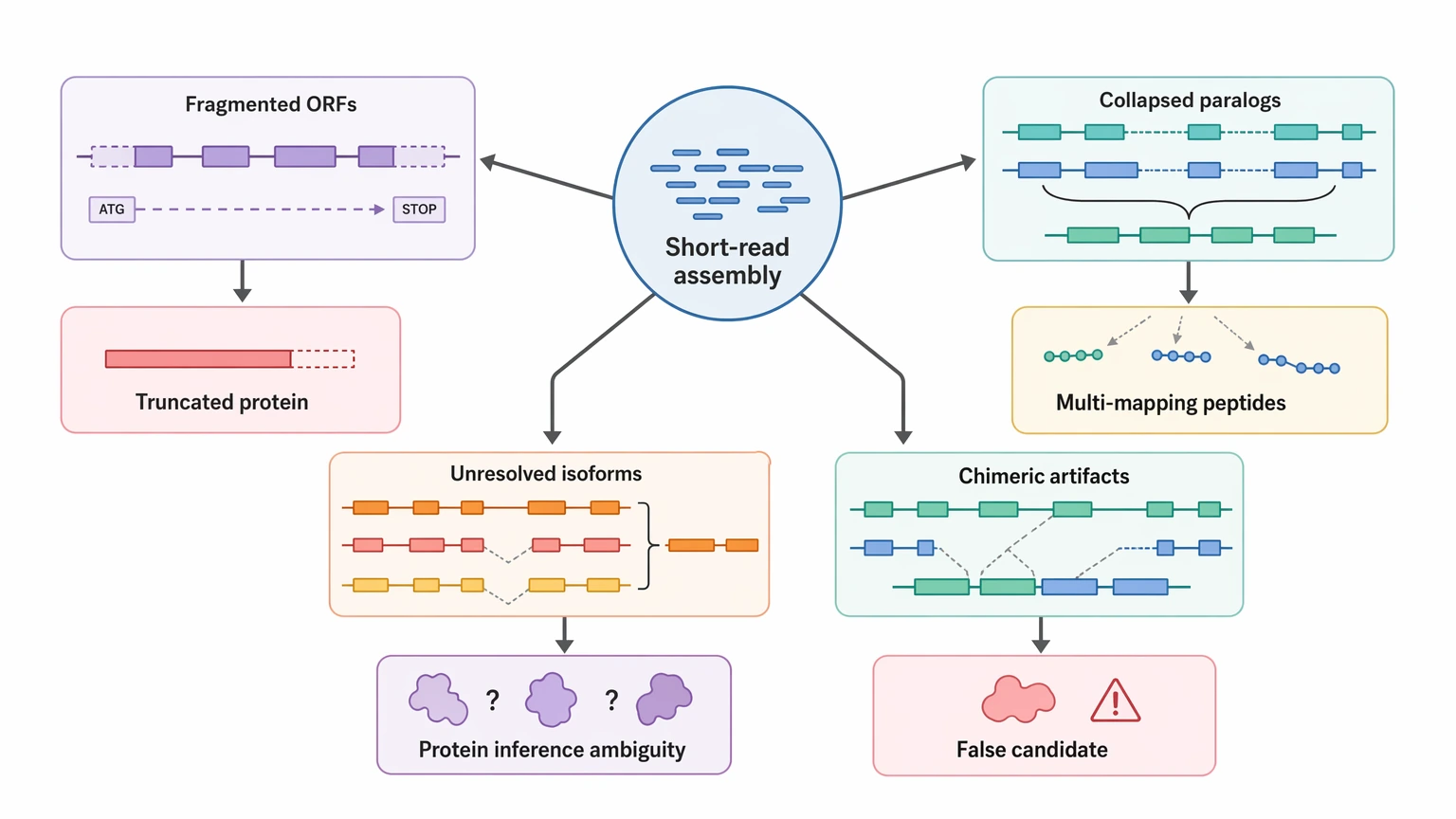
Where Short-Read Assemblies Create Protein-Level Ambiguity
The main weakness of a short-read-only design is structural uncertainty. A short-read assembly may recover many transcripts and still miss the ones that matter most for protein inference.
Three failure modes matter most here:
Fragmented ORFs
A real transcript may show up as multiple partial contigs. When that happens, the predicted protein can be truncated, split, or missing the region needed to explain MS evidence.
Collapsed paralogs or unresolved isoforms
Closely related transcripts can merge into one model or remain difficult to separate. That directly weakens unique peptide mapping and makes it harder to tell whether one peptide supports one protein or several plausible candidates.
Chimeric assembly artifacts
Assembly errors can create transcript models that look complete but do not match a real biological sequence. In downstream proteomics, that can misdirect candidate prioritization more than an obviously partial contig would.
These problems matter most when the project is trying to identify unknown peptides or proteins, not simply catalog transcripts.
When a Hybrid Assembly Changes the Decision
A hybrid assembly adds long-read sequencing to improve transcript continuity while keeping short-read support for coverage and correction. It is most useful when transcript structure changes the meaning of the protein result.
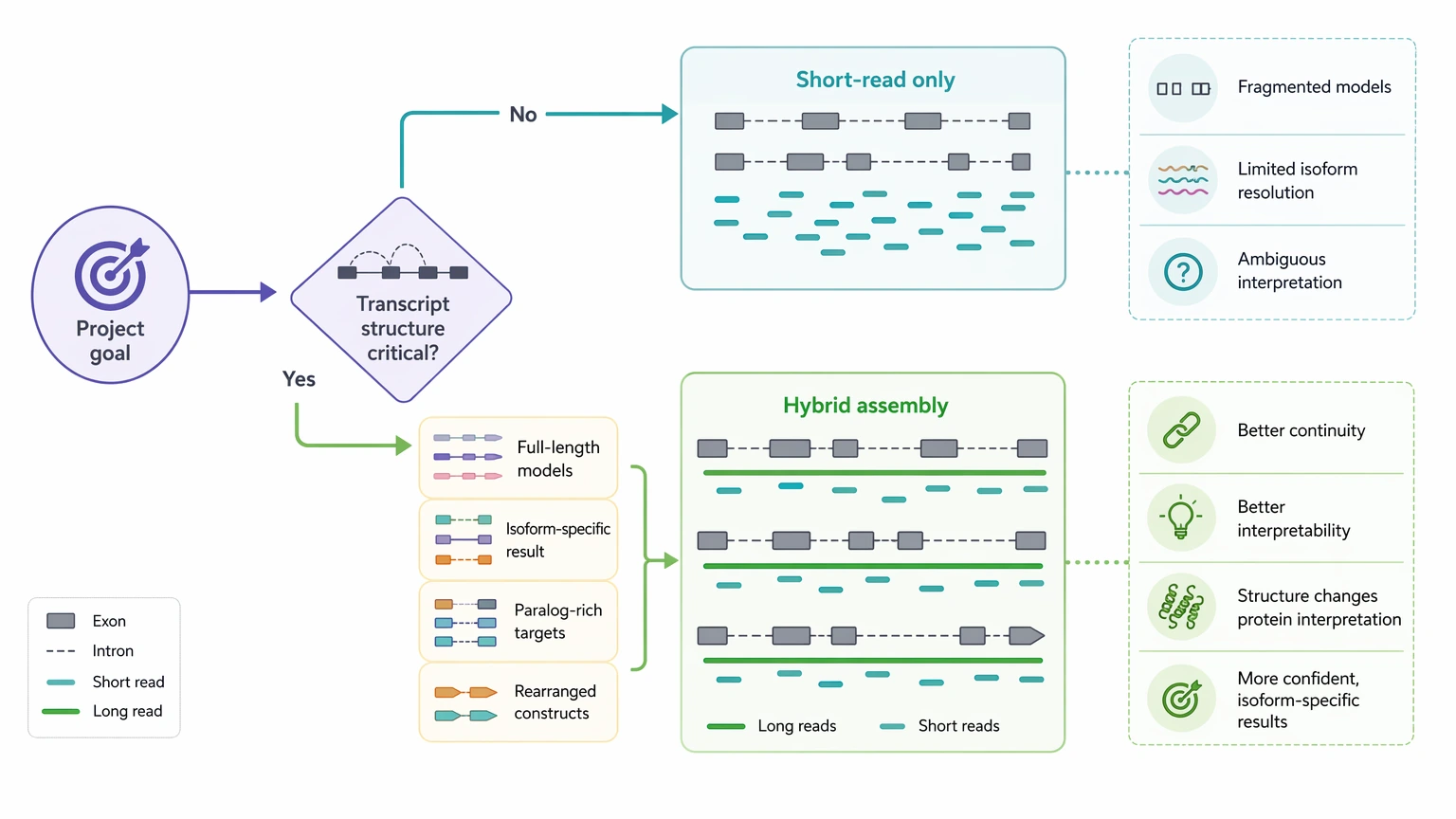
The clearest reasons to escalate are usually these:
In those settings, long reads do not erase uncertainty, but they often improve isoform resolution, ORF completeness, and transcript-to-peptide interpretability enough to justify the extra design complexity.
Service Routes to Consider
For this project scenario, readers usually compare these service routes before requesting a quote or submitting samples.
Side-by-Side Workflow Comparison
The table below focuses on the deliverable that matters most: interpretable protein candidates for LC-MS/MS follow-up.
| Workflow | Best fit | Main strength | Main limitation | Best checkpoint |
|---|---|---|---|---|
| Short-read sequencing only | Moderately complex transcriptomes with broad CDS recovery goals | Efficient candidate-space expansion | May leave partial ORFs or ambiguous transcript structure | Review full-length ORF recovery and peptide mapping uniqueness |
| Short-read first, then escalate | Teams unsure whether transcript structure will be limiting | Preserves a decision checkpoint before adding long reads | May delay final interpretation if escalation becomes necessary | Reassess after initial transcriptome assembly and ORF prediction |
| Hybrid assembly | Isoform-rich, paralog-rich, structurally complex, or novelty-driven projects | Better transcript continuity and structural interpretation | Requires enough sample and still needs downstream confirmation | Compare transcript structure gains against actual protein inference improvement |
The practical takeaway is straightforward: hybrid assembly adds the most value when assembly structure, not just sequence presence, determines whether the protein story is interpretable.
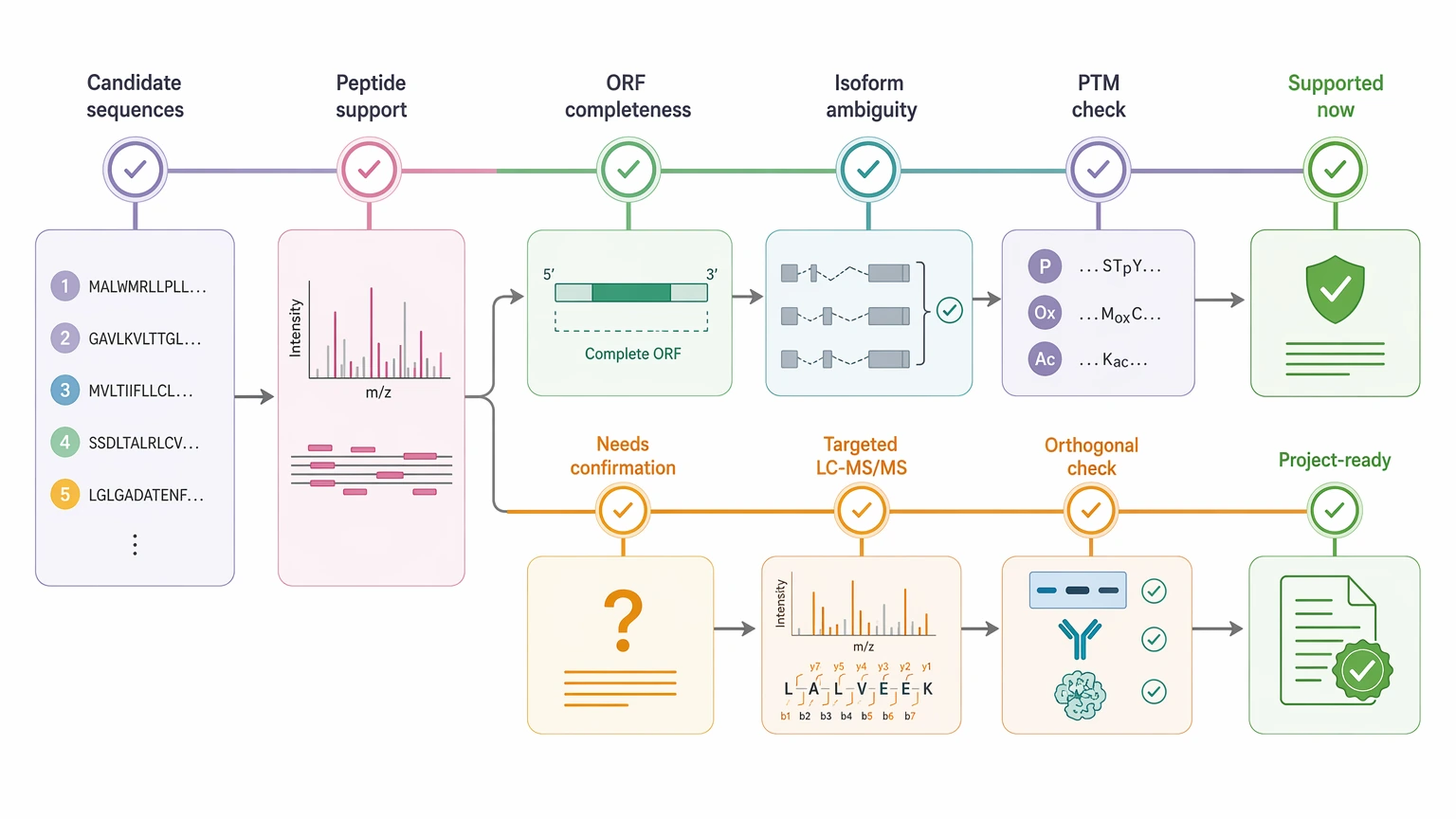
Expected Results and Validation Workflow
Researchers should expect different deliverables from short-read-only and hybrid designs, but neither approach should be treated as final proof by itself.
Immediate deliverables usually include:
Follow-up confirmation is a separate step. It may include targeted LC-MS/MS, orthogonal sequencing checks, reinspection of difficult peptide-spectrum matches, or functional confirmation of the most important candidates.
A useful validation workflow asks two different questions. First, which sequence candidates are supported now? Second, which claims still need confirmation before a sequence is treated as project-ready? That distinction matters even more when MS/MS interpretation uncertainty, PTMs, or incomplete assemblies leave more than one plausible sequence explanation. In PTM-rich cases, modified spectra may support a region while still leaving the exact unmodified sequence context unresolved.
If your team needs help deciding whether a short-read assembly will produce a usable protein candidate handoff, you can submit your requirements to MtoZ Biolabs for project evaluation around transcript complexity, expected deliverables, and the most appropriate validation workflow.
Key Cautions and Practical Limits
A comparison article like this is only useful if it is clear about where each design can fail.
Sample quality or amount limits: poor RNA integrity, biased transcript representation, or low input can reduce both assembly quality and confidence in downstream CDS recovery.
Controls and repeat expectations: ambiguous novel-sequence claims are more convincing when the same candidate appears across replicates, orthogonal preparations, or targeted follow-up runs.
Batch or contamination risk: mixed samples, environmental carryover, and index-related confusion can create apparent novelty that does not hold up under review.
Interpretation boundaries: a predicted protein from transcriptome assembly is still a model. Even with good LC-MS/MS support, transcript evidence and peptide evidence may not uniquely identify one final sequence.
When another method is the better next step: if the study requires exact junction confirmation, full-length variable-region resolution, or direct protein-level sequence proof beyond transcript-supported inference, targeted MS, orthogonal sequencing, or a dedicated protein sequencing strategy may be more informative than adding more short-read depth alone.
Practical Escalation Rules Before You Commit Budget
A staged decision is often the most defensible one.
Start with short-read sequencing when the main goal is broad CDS discovery, the transcriptome is not expected to be highly isoform-rich, and the team can tolerate some incomplete ORFs during first-pass candidate generation.
Escalate to hybrid assembly when early review shows repeated multi-mapping peptides, high-value targets with partial ORFs, unresolved splice alternatives, or candidate proteins that differ mainly in transcript structure.
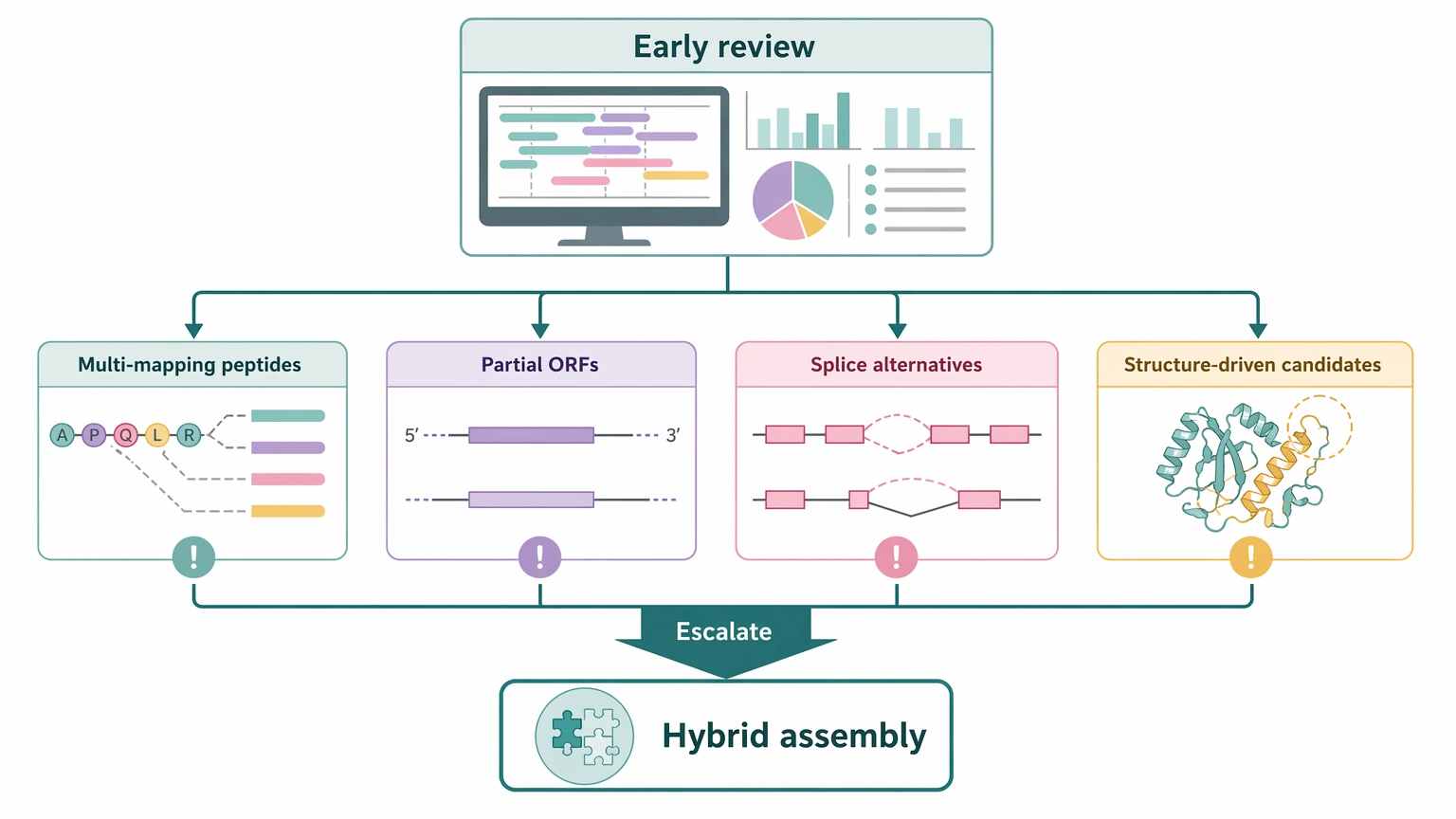
Keep that review centered on the protein decision. A transcriptome can look acceptable overall and still be weak for the few transcripts that matter most to the LC-MS/MS readout.
A practical mid-project checkpoint is to ask whether the current assembly supports:
Technical Summary and Consultation Guidance
For de novo peptide and protein discovery, short-read sequencing is often adequate when the immediate objective is broad de novo assembly support for CDS recovery and candidate filtering, not exact structural resolution of every transcript. A hybrid assembly is more appropriate when assembly contiguity, isoform resolution, or paralog separation directly affects protein inference from LC-MS/MS. This matters most in non-model organisms, secreted peptide studies, engineered constructs, and other projects where missing transcript structure can carry uncertainty into candidate sequence interpretation. If that matches your study, contact us and evaluate your project with MtoZ Biolabs by sharing sample type, RNA condition, current LC-MS/MS findings, expected candidate outputs, and the level of sequence confidence needed for downstream decisions.
FAQ
Can low-abundance transcripts still justify hybrid assembly even if the transcriptome is not very complex?
Yes. If the priority targets are rare and structurally important, a simple overall transcriptome profile does not guarantee that those specific transcripts will assemble in a protein-useful way.
Does a better N50 automatically mean better protein inference?
No. A stronger contiguity metric can be encouraging, but protein inference depends more directly on CDS completeness, correct ORF structure, and whether peptides map uniquely to the predicted proteins that matter.
Should long-read sequencing be added just because database search coverage is poor?
Not by itself. Poor database coverage may reflect novelty, PTMs, incomplete references, or weak peptide uniqueness. Long reads help most when transcript structure is the missing piece in the interpretation.
What is the most useful checkpoint after a short-read pilot?
Review the top candidates for full-length ORF recovery, unique peptide mapping, and transcript models that still leave more than one plausible protein assignment.
When is transcript evidence less helpful than direct protein-focused confirmation?
When the project depends on exact mature peptide sequence, junction placement, variable-region identity, or PTM-localized interpretation that assembled transcripts cannot resolve with enough confidence.
How to order?

