





LC-MS/MS Protein Sequencing: Workflow, De Novo Interpretation, and Proteomics Applications
-
LC-MS/MS protein sequencing usually relies on peptide-level analysis after enzymatic digestion rather than direct intact-protein readout.
-
LC reduces sample complexity before MS, while MS/MS fragmentation provides sequence-defining fragment ions.
-
Database search is efficient when reference sequences exist, whereas de novo sequencing helps when reference coverage is incomplete or unknown.
-
Sequence coverage, PTMs, protease choice, and data quality all influence how confidently proteins can be reconstructed.
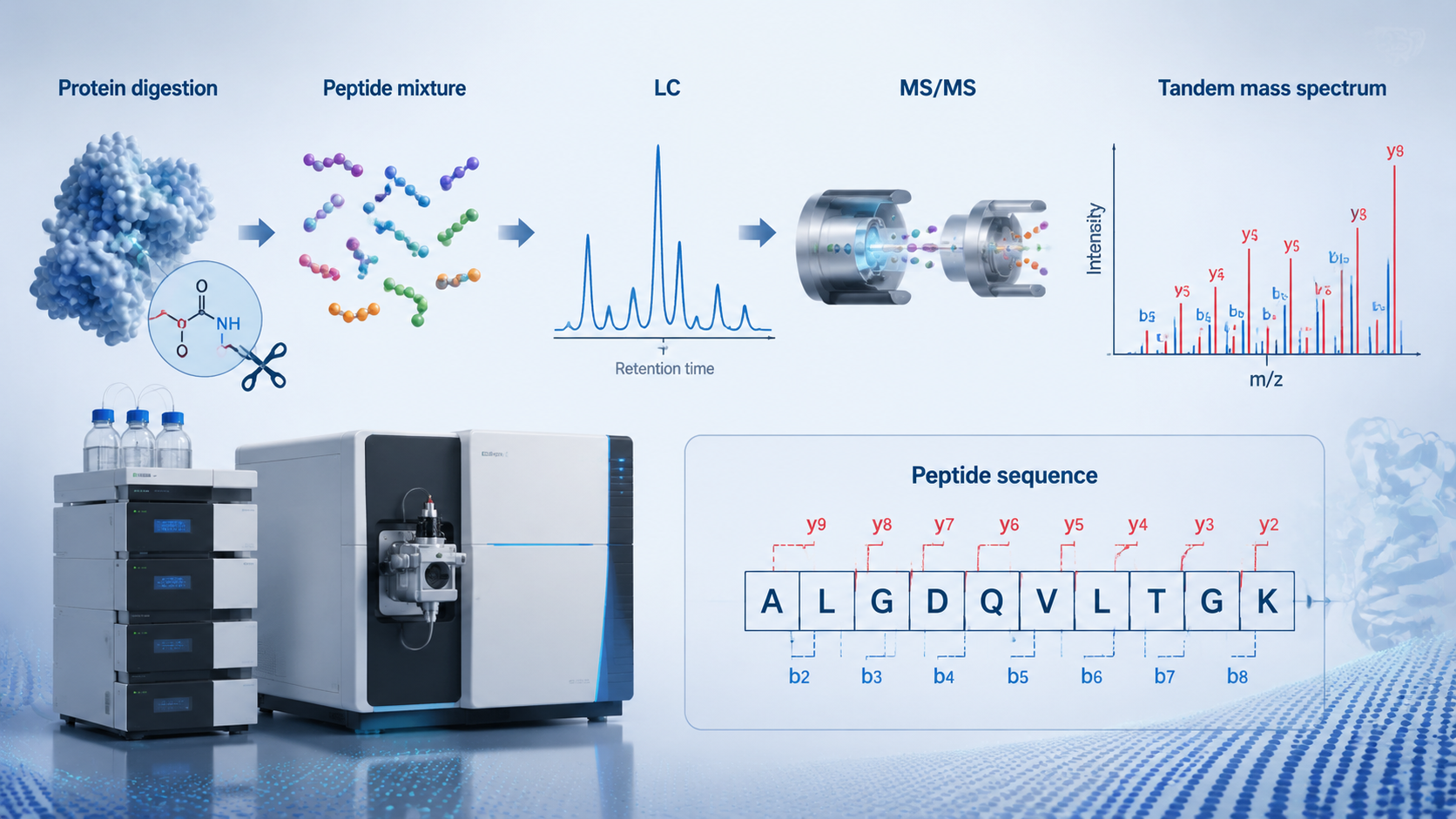
LC-MS/MS protein sequencing combines chromatographic peptide separation with tandem mass spectrometry to identify proteins, reconstruct peptide sequences, and support quantitative proteomics in complex biological samples. In practice, most workflows do not sequence whole intact proteins residue by residue from scratch. Instead, they digest proteins into peptides, separate those peptides by LC, fragment them by MS/MS, and infer peptide or protein identity through database matching, de novo interpretation, or hybrid approaches.
Key Takeaways
What Does LC-MS/MS Protein Sequencing Actually Do?
In bottom-up proteomics, proteins are extracted, digested into peptides, and analyzed as peptide ions rather than as whole proteins. The LC stage separates peptides over time, which makes the MS stage more sensitive and interpretable. The MS/MS stage fragments peptide precursors and records the resulting ion series, allowing software or manual review to infer amino acid order, PTMs, and peptide identity.
Related Services
Mass Spectrometry Protein Sequencing Service
Mass Spectrometry-Based Protein Sequencing Service
Mass Spec Protein Sequencing Service
De Novo Peptide Sequencing Services
De Novo Peptide Sequencing Service
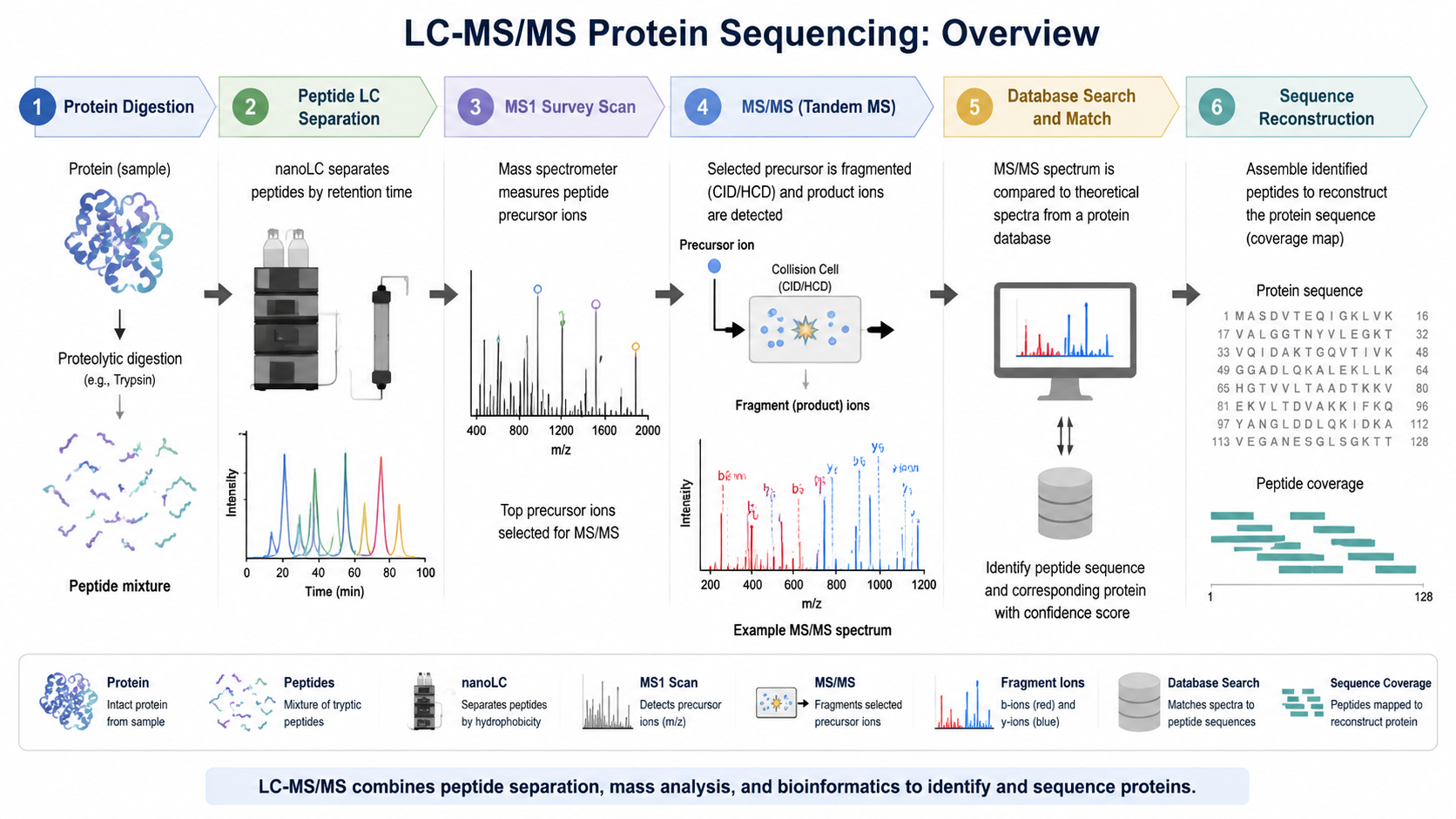
Core LC-MS/MS Sequencing Workflow
The workflow starts with protein extraction, cleanup, and proteolytic digestion, most often with trypsin. The peptide mixture is then injected into reversed-phase liquid chromatography, which separates peptides by hydrophobicity and reduces co-elution. As peptides enter the mass spectrometer, MS1 scans measure precursor mass-to-charge values, and selected precursors are fragmented to produce MS2 spectra.
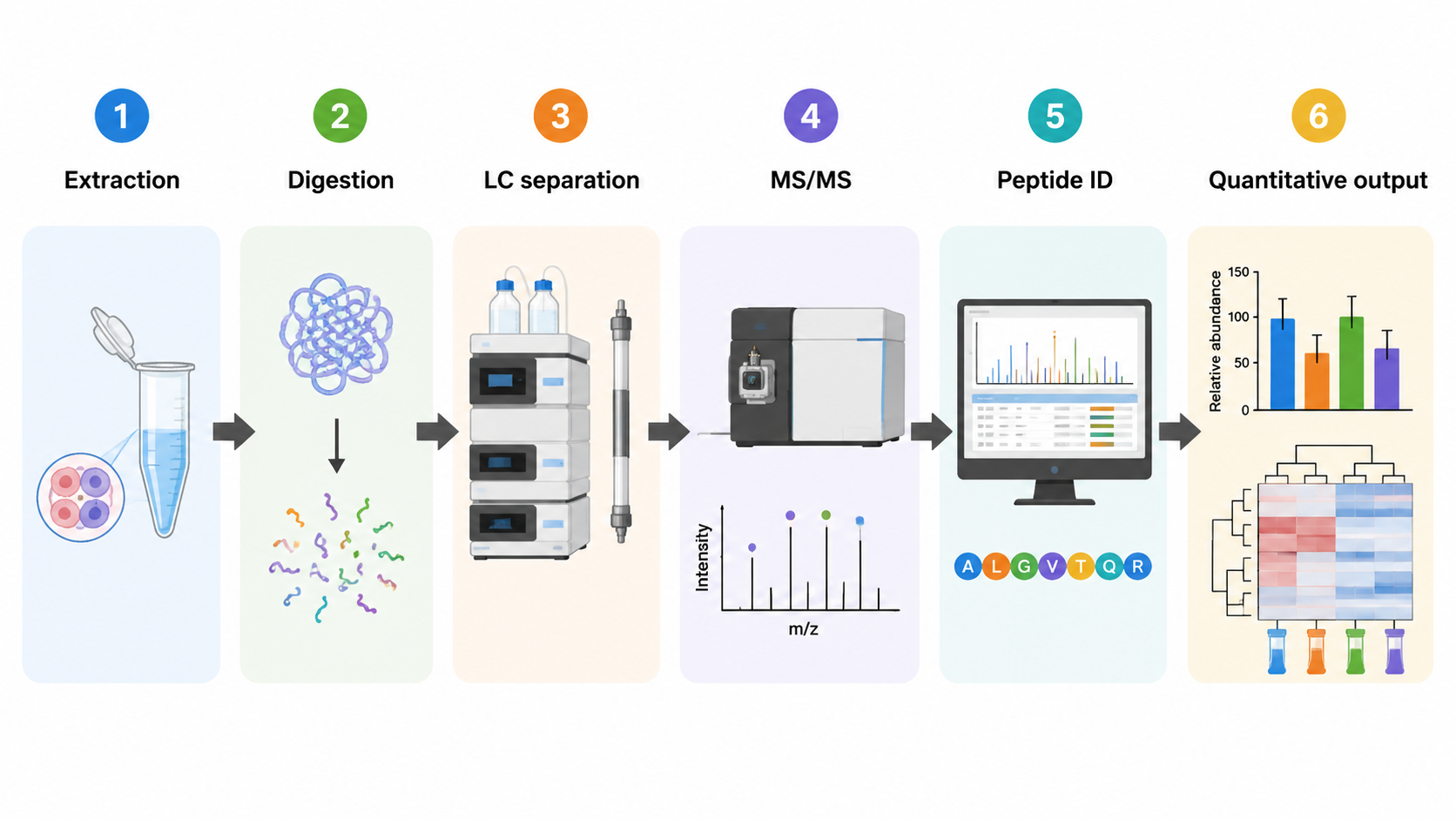
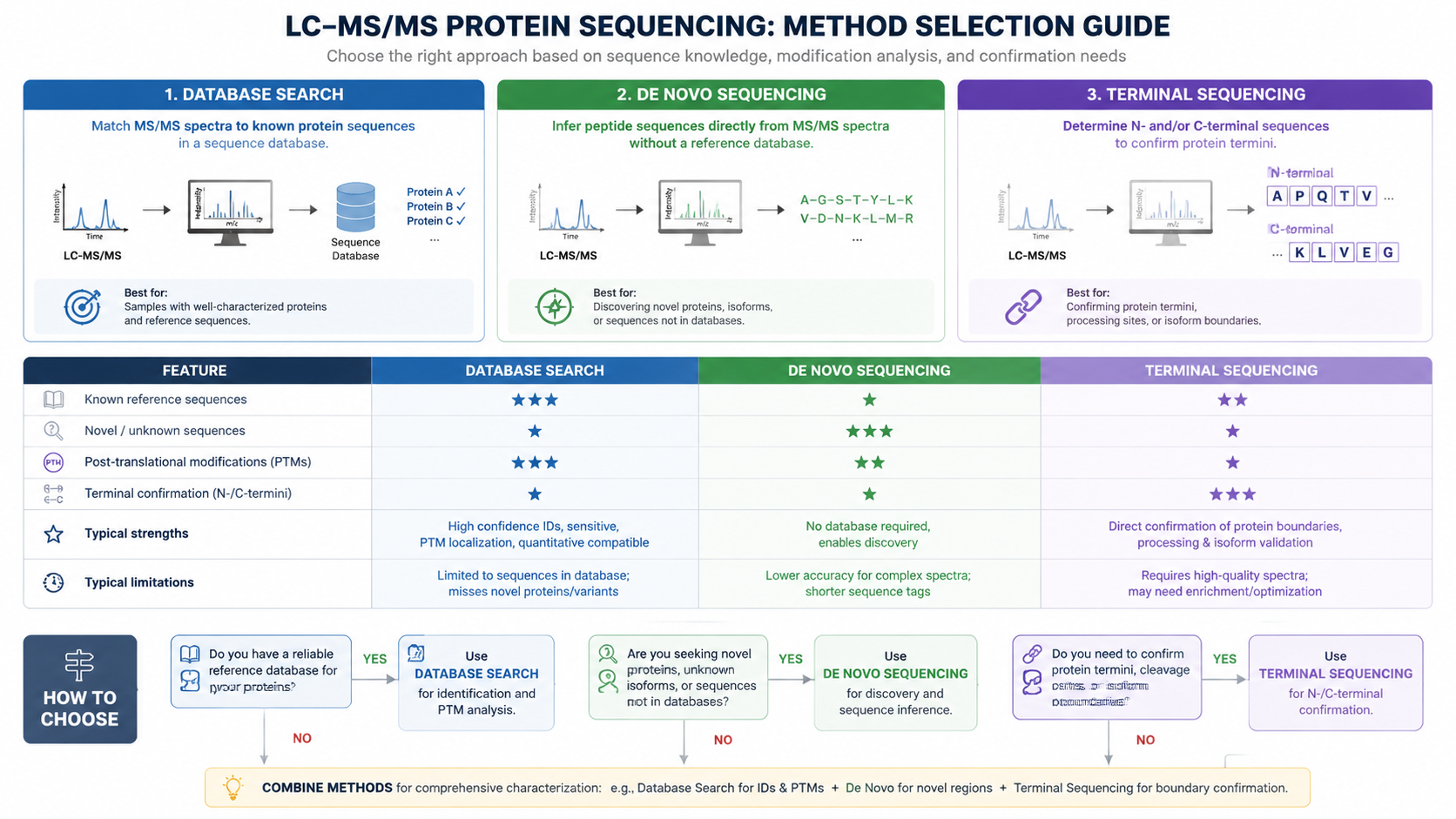
Database Search Versus De Novo Sequencing
1. Database Search
Database search works well when the organism or target protein family is known. Spectra are compared with theoretical peptide candidates generated from a reference database.
2. De Novo Interpretation
De novo sequencing is useful when reference databases are incomplete, when novel peptide variants are expected, or when antibody, venom, microbial, or environmental samples may contain uncharacterized sequences.
Strengths and limitations
| Strength | Why It Matters? | Limitation | Practical Consequence |
|---|---|---|---|
| High sensitivity | Detects low-abundance peptides | Sample prep complexity | Errors upstream can reduce coverage |
| Broad throughput | Profiles many proteins at once | Partial sequence coverage | Not every protein is reconstructed end-to-end |
| PTM detection | Reveals modifications and site evidence | Complex data analysis | Requires strong software and review |
| Flexible applications | Works across many fields | Database dependence | Unknown sequences may need de novo support |
Common Applications
LC-MS/MS protein sequencing is widely used in disease mechanism research, biomarker discovery, drug target studies, food allergen analysis, agricultural trait studies, and environmental protein characterization.
Practical Method Selection
Use routine database search when high-throughput identification is the main goal. Add de novo analysis when sequence novelty is plausible. Consider Edman or terminal sequencing when clean N- or C-terminal confirmation is required.
FAQ
1. What is LC-MS/MS protein sequencing?
It is a workflow that separates peptides by liquid chromatography, fragments them by tandem mass spectrometry, and interprets the resulting spectra to identify peptide and protein sequences.
2. Does LC-MS/MS read the full protein directly?
Usually no. Most proteomics workflows use digested peptides and infer protein identity from peptide evidence, although top-down methods can analyze intact proteins in specialized cases.
3. When is de novo sequencing necessary?
De novo sequencing is most useful when reference sequences are incomplete, when novel variants are expected, or when direct spectrum-based sequence reconstruction is needed.
Conclusion
LC-MS/MS protein sequencing is one of the central analytical engines of modern proteomics because it combines separation, sensitivity, and spectrum-based sequence interpretation in one scalable workflow. The most informative studies align sample preparation, fragmentation strategy, database context, and de novo logic to match the biological question instead of treating sequencing as a one-size-fits-all pipeline.
How to order?

