





Hybridoma Antibody Sequencing: How to Recover VH/VL Sequences From Legacy Clones
- restore a clone by recombinant re-expression
- document identity for archive or patent cleanup
- transfer a program after records were lost
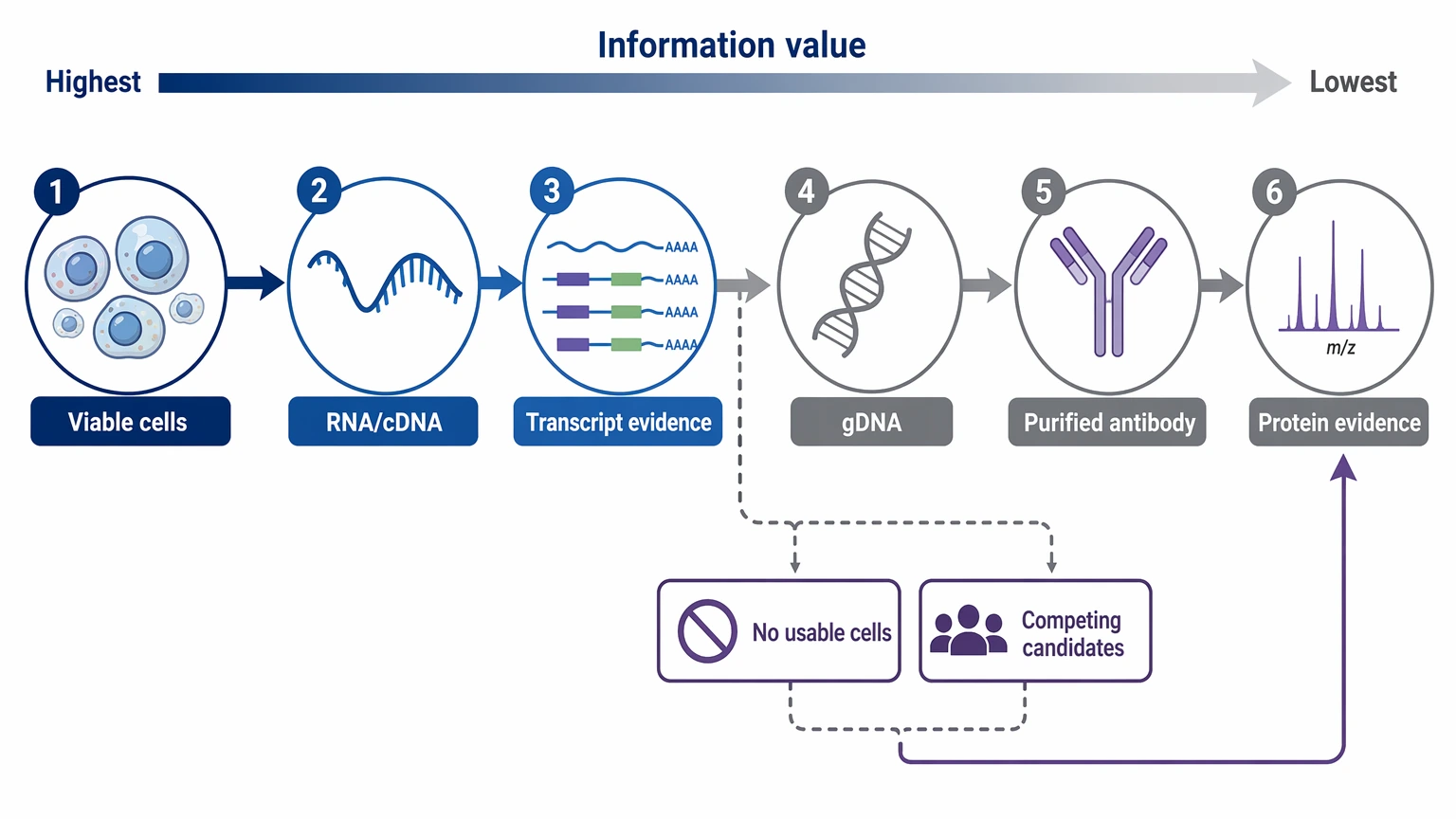
- viable hybridoma cells
- frozen cell pellets
- extracted RNA
- archived cDNA
- genomic DNA
- culture supernatant
- purified monoclonal antibody
- historical isotype notes
- previous fragment data or clone-specific PCR products
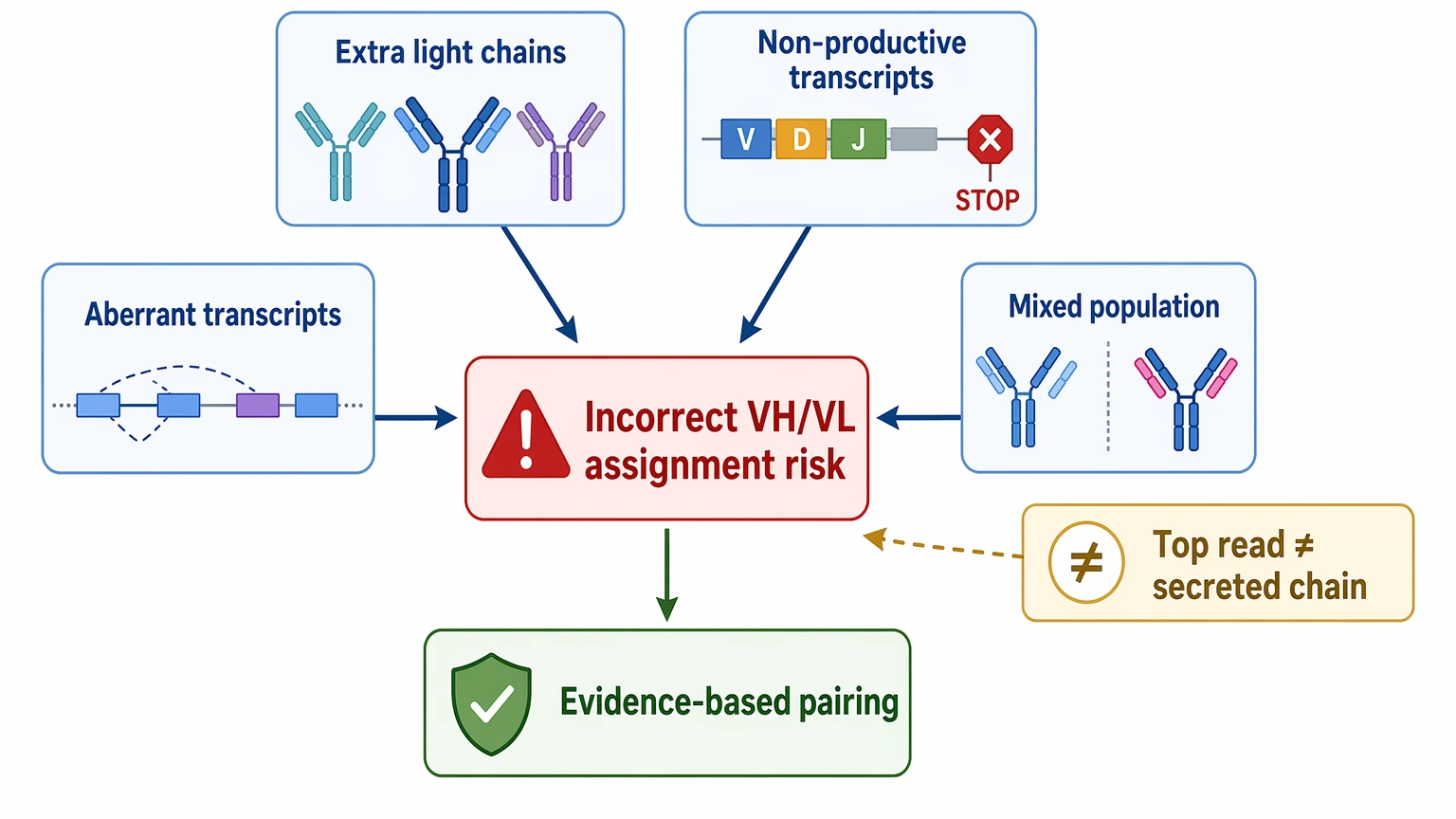
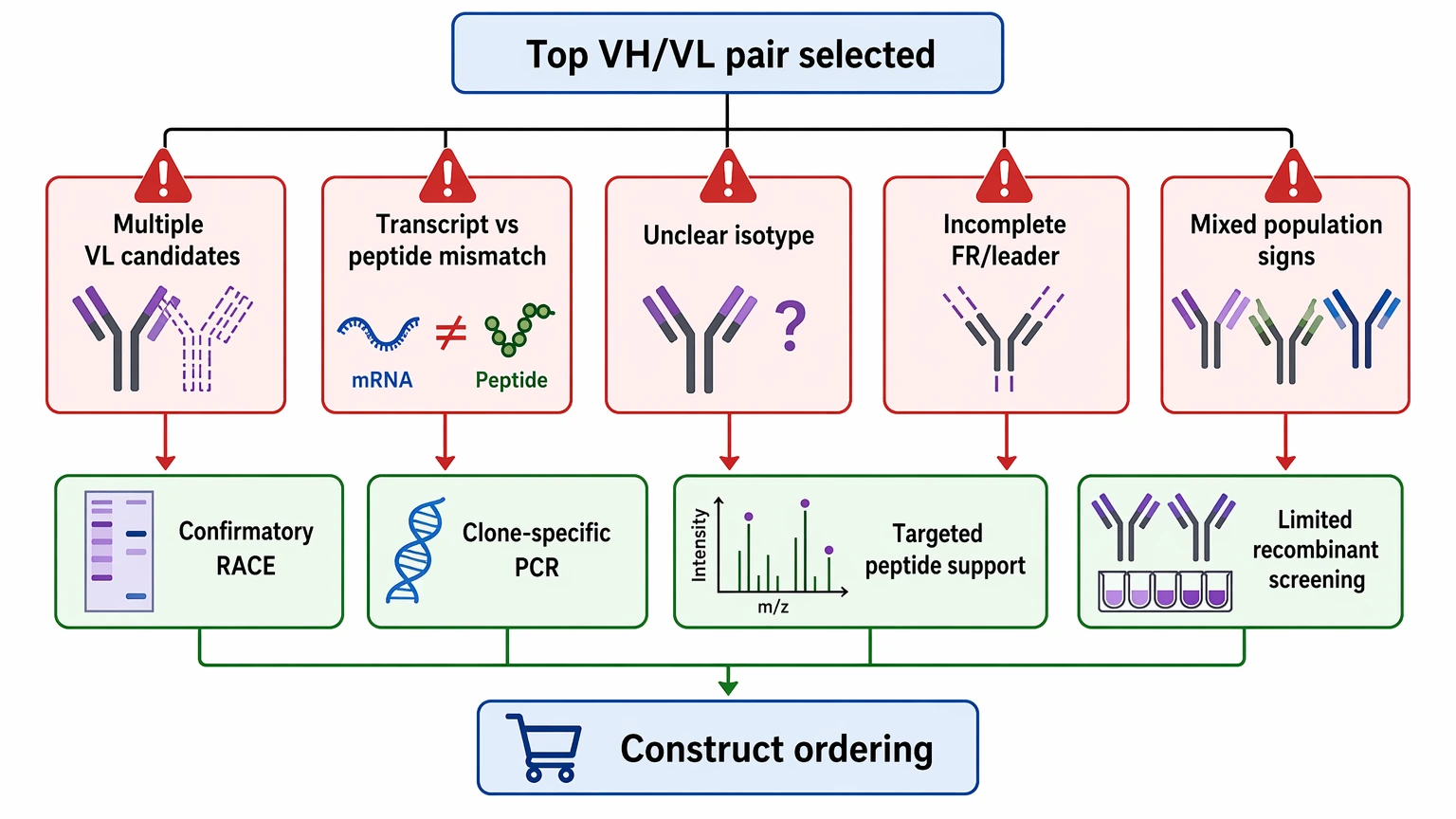
- more than one plausible VL sequence
- mismatch between transcript abundance and peptide support
- unclear isotype or light-chain class
- incomplete framework region or leader sequence
- signs of mixed population or clone instability
- historical assay data from poorly documented passages
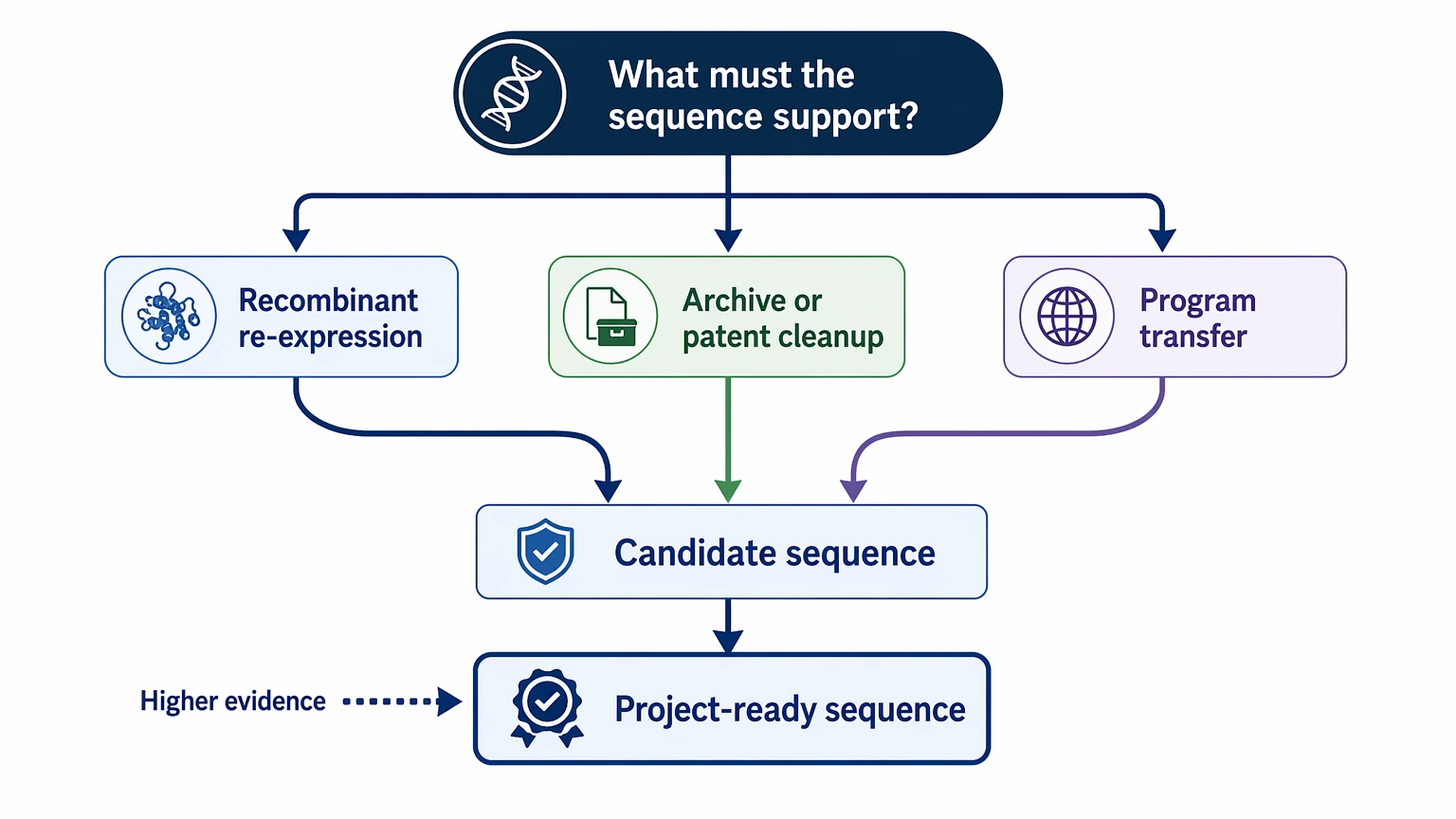
- Candidate identification: one or more plausible VH sequence and VL sequence candidates are found.
- Defensible sequence set: the selected candidates are supported by productive rearrangement analysis, framework region continuity, and sample context.
- Project-ready sequence: the final pair is strong enough to support recombinant re-expression planning, with orthogonal sequence validation and a defined binding concordance check.
If you still have viable or frozen hybridoma cells, the best starting point for hybridoma antibody sequencing is usually RNA converted to cDNA, followed by RACE or targeted amplification. In most cases, that route gives you the clearest shot at a usable VH sequence and VL sequence because it can recover full variable regions and support interpretation of productive rearrangement and chain pairing. If the cells are gone and only secreted antibody is left, antibody protein sequencing by LC-MS/MS with de novo peptide sequencing becomes the fallback. Even then, protein-derived sequence candidates usually need extra validation before you treat them as ready for recombinant re-expression.
The deciding question is not whether you can generate sequence-like output at all. What really matters is whether you can recover a heavy chain variable region and light chain variable region that can be tied back to the legacy hybridoma clone, fit the expected isotype, and are documented well enough for archive repair, patent support, or clone replacement.
Where Legacy Hybridoma Rescue Usually Gets Stuck
Most rescue projects start with a useful monoclonal antibody and incomplete records. A team may still have historical binding data, assay performance results, or in vivo data linked to a clone name, but no reliable sequence file. Sometimes a frozen vial still exists. Sometimes all that remains is a cell pellet, old RNA, culture supernatant, or purified antibody.
That gap creates two practical problems. First, the project cannot move cleanly into recombinant re-expression, portfolio transfer, or sequence-based documentation. Second, the technical risk is easy to underestimate. A partial CDR call, a dominant transcript, or a peptide-supported reconstruction can look persuasive on paper and still fall short when you need to choose the correct VH sequence and VL sequence.
Legacy rescue becomes more difficult as the material degrades or disappears. Once a thawed vial loses viability, RNA integrity drops, or the last purified antibody aliquot is used up, the best recovery routes narrow fast. That is why sample triage should happen before exploratory work consumes limited material.
The Main Reasons Sequence Recovery Stays Uncertain
For this kind of project, four cause categories usually drive most of the uncertainty.
1. The Starting Material Sets the Confidence Ceiling
Not every archived input can support the same answer. Viable cells and intact RNA can support full heavy chain variable region and light chain variable region recovery, including leader sequence and framework region coverage. Purified antibody can support antibody protein sequencing, but it does not directly show the transcript landscape inside the original hybridoma.
2. A Legacy Hybridoma Clone May Not Produce Only One Clean Chain Pair
Hybridomas can carry extra light chains, non-productive transcripts, aberrant transcripts, or mixed populations caused by instability. The most abundant read is not always the secreted functional chain. That is why dominant transcript detection is weaker than evidence-based chain pairing.
3. Damaged Nucleic Acid Limits Full-Length Recovery
Poor RNA integrity often prevents clean recovery of complete variable regions. Short fragments may still amplify, but missing framework region or leader sequence coverage makes downstream construct design less secure. Genomic DNA can reveal a V(D)J rearrangement, yet by itself it does not show whether that rearrangement was the productive rearrangement driving secretion.
4. The Validation Standard Is Often Too Low for the Intended Use
A candidate sequence list may be enough for archive notes. It usually is not enough for recombinant re-expression. If the recovered sequence will be used to design constructs or support formal documentation, the standard should include productive rearrangement assessment, pairing logic, and at least one orthogonal check such as confirmatory RACE, clone-specific PCR, LC-MS/MS peptide support, or binding concordance after re-expression.
Project-Planning Guide: How to Choose the Right Recovery Route
This article addresses a project-planning problem. The best workflow is not a generic lab sequence. It starts with the material you still have, the decision you need to make next, and the level of evidence that decision requires.
Step 1: Define What the Sequence Must Support
Before sequencing starts, put the downstream use into one sentence. Are you trying to:
This matters because “candidate sequence identified” and “project-ready sequence selected” are not the same outcome. If the next step is construct design, the evidence bar is higher than it is for internal documentation alone.
Step 2: Rank All Surviving Materials by Information Value
Build a full inventory before choosing a method. Useful inputs may include:
For most recombinant re-expression projects, cell-derived nucleic acid ranks first because it can provide transcript-level evidence and help separate productive rearrangement from background immunoglobulin signals. Protein-derived evidence becomes more important when no usable cells remain or when transcript results leave you with several competing candidates.
If you have several partial sample types and need to conserve material, submit your requirements and sample inventory for a workflow review before you commit to one route. In that setting, MtoZ Biolabs can evaluate whether the project should begin with cDNA-based recovery, antibody protein sequencing, or a combined confirmation strategy.
Step 3: Match the Sequencing Route to the Sample State
The sample state usually tells you what the best first move is.
| Starting material | Preferred route | What it can support well | Main limitation |
|---|---|---|---|
| Viable hybridoma cells | RNA to cDNA, then RACE or targeted amplification | Full VH sequence and VL sequence candidates, transcript interpretation | Requires recoverable cells |
| Frozen cell pellet | RNA extraction first, genomic DNA as backup | Good variable-region recovery if RNA integrity is still usable | Degraded RNA can reduce full-length coverage |
| Extracted RNA or cDNA | Targeted amplification and confirmatory RACE | Direct transcript recovery without reviving cells | Prior degradation or primer bias may limit interpretation |
| Genomic DNA | V(D)J rearrangement analysis plus confirmation | Useful fallback when RNA fails | Productive status and chain pairing are less direct |
| Supernatant or purified antibody | Antibody protein sequencing by LC-MS/MS and de novo peptide sequencing | Protein-only rescue when cells are gone | Exact reconstruction and pairing may remain ambiguous |
When cells or RNA are available
This is usually the strongest route. cDNA-based recovery can capture full variable-region information and often the leader sequence. It is also the best place to start if you need to identify a productive rearrangement and determine whether more than one plausible light chain is present.
When only genomic DNA survives
Genomic DNA can still reveal candidate V(D)J rearrangement patterns, but it is better treated as a rescue-support route than as the sole basis for expression-ready chain assignment. It becomes much more convincing when paired with other context, such as known isotype or peptide evidence.
When only antibody remains
If no viable cells or usable nucleic acid survive, antibody protein sequencing is the practical fallback. LC-MS/MS and de novo peptide sequencing can recover large parts of the variable regions, but some residue calls and heavy/light attribution may still need interpretation. In protein-only projects, confidence improves when sequence candidates also fit the known isotype, expected framework region architecture, and later binding concordance.
Step 4: Set the Minimum Evidence for a Defensible VH/VL Pair
Set this standard before the data arrive. A usable output for recombinant re-expression should usually include the following evidence.
| Evidence checkpoint | Why it matters |
|---|---|
| Full or near-full heavy and light variable-region coverage | Supports construct design rather than fragment-level inference |
| Productive rearrangement identified | Excludes obvious nonfunctional candidates |
| Credible chain pairing logic | Reduces the chance of combining the wrong heavy and light chains |
| Isotype consistency | Supports clone attribution |
| Framework region and leader sequence context where possible | Improves interpretation and expression planning |
| Orthogonal sequence validation | Strengthens final sequence selection |
| Binding concordance plan | Confirms whether the recombinant product behaves like the historical antibody |
If your team is deciding whether to move ahead with reformatting, this is also the point to request a project review. A practical consultation can help define whether your current data package is enough for recombinant re-expression or whether another validation layer should come first.
Step 5: Do Not Treat the Top Sequence Pair as Final Too Early
Many rescue projects go off track after sequencing, not during sequencing. The common mistake is moving straight from top-ranked candidates to construct ordering without resolving obvious warning signs.
Pause and add confirmation if you see:
In those cases, another round of confirmatory RACE, clone-specific PCR, targeted peptide support, or limited recombinant screening of the top chain pairs is often more informative than repeating the same initial assay.
What “Project-Ready” Actually Means
A recovered sequence becomes project-ready only when the evidence matches the decision you need to make. In practice, results usually fall into three tiers:
That distinction matters even more in archive repair and patent support. A report should clearly separate candidate sequence identification from functionally supported final sequence selection.
Practical Submission Notes Before You Start
Prepare more than the sample tube. For a legacy hybridoma clone, the most useful background usually includes sample type, storage history, known isotype, whether the clone was ever unstable, the intended downstream use, and any surviving assay that can be used to compare recombinant and historical binding.
Avoid pooling different archived vials or antibody lots unless you already know they are equivalent. Separate handling can expose drift, relabeling, contamination, or mixed-chain behavior that would otherwise stay hidden.
Conclusion
Hybridoma antibody sequencing works best when the recovery route follows the material that still survives and the validation plan matches the next project decision. Cell-derived RNA converted to cDNA is often the strongest starting point because it can support full heavy chain variable region and light chain variable region recovery, productive rearrangement assessment, and stronger chain pairing logic. When only secreted antibody remains, antibody protein sequencing can still recover useful sequence evidence, but protein-derived candidates usually need more confirmation before they can replace the original clone. For legacy rescue projects involving frozen cells, pellets, uncertain RNA, or protein-only archives, contact MtoZ Biolabs to discuss the sample state, evaluate your project, and define what level of sequence validation is appropriate before moving into recombinant re-expression or formal documentation.
FAQ
Can a non-productive rearrangement still appear convincing in sequencing data?
Yes. A non-productive rearrangement may still be amplified or detected at appreciable levels, especially in nucleic-acid-based workflows. That is why in-frame status, stop codons, and plausible framework region structure should be checked before treating a sequence as functional.
If two light-chain candidates look plausible, how should the project proceed?
Do not choose by abundance alone. Bring in orthogonal evidence, such as peptide support, clone-specific confirmation, or small-scale recombinant testing of the top heavy/light combinations. That extra step is often less costly than advancing the wrong pair.
Is leader sequence recovery necessary for every rescue project?
Not always for documentation, but it is highly useful for recombinant re-expression planning. Missing leader sequence information can complicate construct design and make it harder to judge whether the recovered variable region is complete.
Can purified antibody alone support a patent or archive cleanup effort?
It can support sequence reconstruction, but the strength of that support depends on coverage and ambiguity. Protein-only evidence is often better treated as a clearly labeled candidate sequence package unless additional confirmation is available.
What background records are most useful to provide with the sample?
The most helpful records are known isotype, species, approximate clone age, storage history, any prior sequencing fragments, and one assay that can later be used for binding concordance testing.
Does a recovered sequence prove the recombinant antibody will match the original hybridoma product?
No. Sequence recovery improves the odds of recreating the original antibody, but equivalence still needs downstream confirmation. Binding concordance is the most practical next check when the recombinant product will replace the historical monoclonal antibody in active research.
How to order?

