





How to Evaluate De Novo Peptide Sequencing Software Before Choosing In-House Analysis or External Interpretation Support
- several near-top candidates with different residue order
- broken b ions and y ions ladders
- a plausible mass shift with weak PTM localization
- unstable sequence tag assignments across fractions or replicates
- disagreement between analysts during manual interpretation
- Do you need a full-length sequence, a robust sequence tag, or a narrowed candidate set?
- Will the result guide synthesis, impurity characterization, or only internal screening?
- Does the next step require orthogonal validation?
- the same candidate recurs across replicate spectra
- the core sequence tag survives preprocessing changes
- alternative candidates differ mainly at known weak regions
- manual interpretation can explain major peaks without forced assignments
- uncertain PTM localization near missing ladder regions
- two or more plausible explanations for the same delta mass
- unresolved isobaric residues
- incomplete support around biologically or chemically important positions
- heavy reliance on manual interpretation to rescue software output
- multiple fractions or runs must be integrated
- ambiguous PTM reasoning drives the final call
- candidate pruning is consuming specialist time
- a wrong sequence would trigger expensive synthesis or repeat studies
- a documented statement on whether current de novo peptide sequencing software is sufficient for this dataset
- a justified ranked candidate list or a bounded consensus sequence
- explicit notes on unresolved residues, termini, or PTMs
- a defined handoff decision for in-house analysis versus interpretation support
- synthetic peptide confirmation
- intact mass consistency
- targeted review of fragment-ion support around uncertain positions
- replicate acquisition or complementary LC-MS/MS data
- another orthogonal check when PTM placement or residue identity remains critical
- Sample quality or amount limits: if material is scarce or impure, repeated internal reprocessing may use up the chance for a better confirmation strategy later.
- Controls, replicates, and repeat expectations: de novo calls are stronger when replicate spectra agree. If repeat acquisition is impossible, uncertainty should be stated more conservatively.
- Batch or contamination risk: carryover, mixed precursors, or fraction cross-talk can create misleading sequence consistency.
- Interpretation boundaries: a software-generated candidate is not the same as confirmed unknown peptide identification. Report unresolved positions directly.
- When another method is the better next step: if the main uncertainty is precursor purity or missing fragmentation, reacquisition or a different fragmentation mode may help more than more software tuning.
- When outside support is the better next step: if your team is spending more time debating annotations than planning validation, the bottleneck is interpretation capacity rather than software access.
Use in-house analysis only when your de novo peptide sequencing software, analyst review, and LC-MS/MS evidence point to the same sequence interpretation with only limited unresolved ambiguity. If top candidates change across replicate spectra, PTM localization remains uncertain, or the sequence repeatedly needs manual rescue after scoring, external interpretation support is often the lower-risk choice before validation or reporting.
Quick decision block
Stay in-house when: the MS/MS spectrum shows interpretable fragment-ion coverage, the top candidate stays stable across replicates, and the next validation step is clear.
Escalate for interpretation support when: candidate sequences conflict, post-translational modification (PTM) reasoning drives the call, or the cost of a wrong sequence is higher than the cost of expert review.
Key limitation: a high software score does not by itself establish full-length sequence confidence, and Leucine/Isoleucine ambiguity or uncertain PTM site placement may still remain unresolved by tandem mass spectrometry alone.
Where Teams Usually Get Stuck
This decision usually comes up after a team has already collected seemingly usable LC-MS/MS data from an unknown peptide, impurity-related fragment, venom peptide, modified peptide, or a novel sequence region affected by a database search limitation. The software returns candidates, but the project still does not have an answer the team can defend.
Typical warning signs include:
At that stage, the question is no longer whether de novo sequencing is possible in principle. The real question is whether the current data and available analyst time can support a sequence call that is credible enough for synthesis, impurity follow-up, or internal reporting.
Why Software Output Often Stops Short
Most in-house versus external-support decisions fall into four main cause categories.
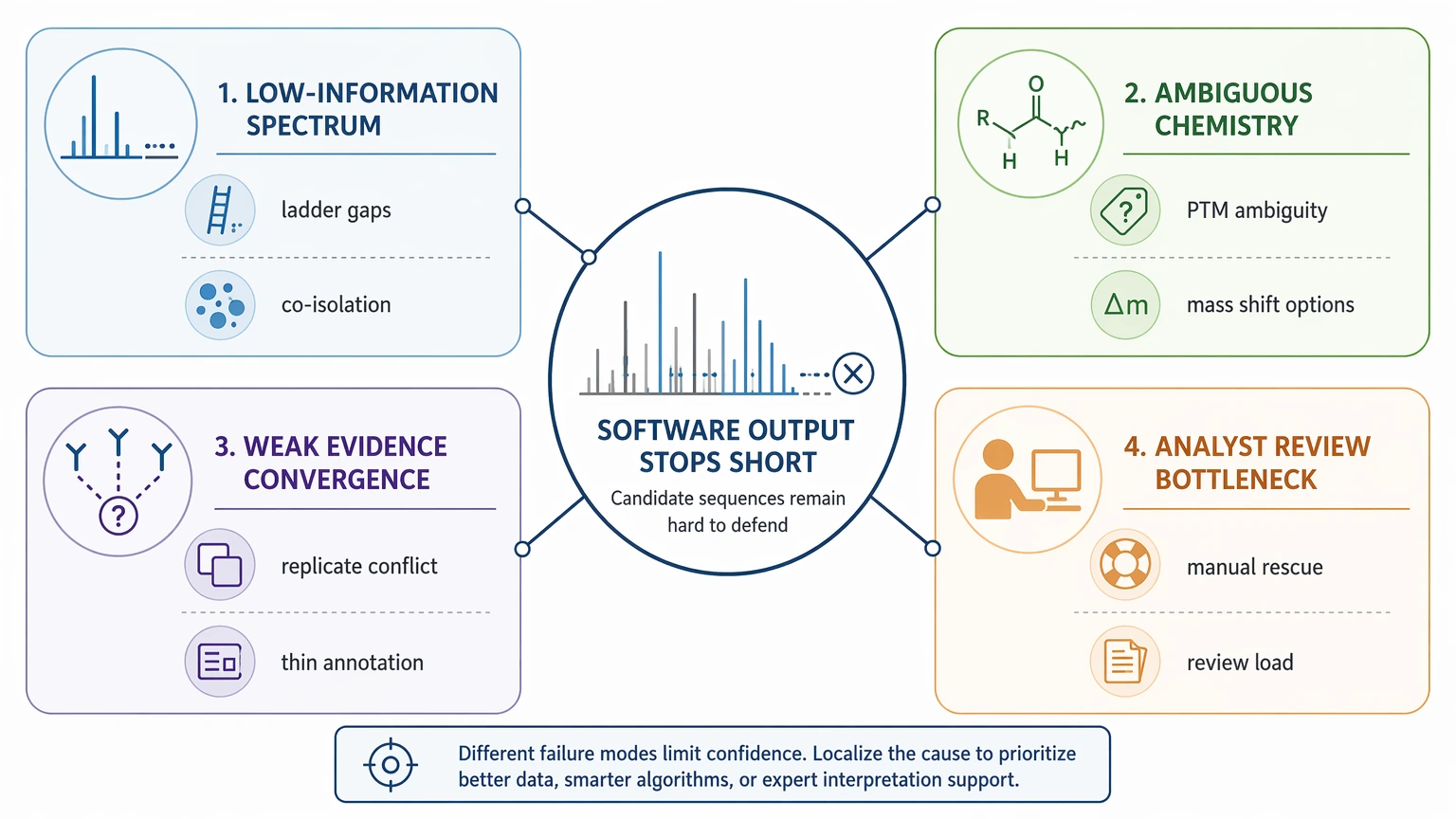
1. The MS/MS spectrum lacks enough sequence information
Some spectra simply do not contain a continuous fragment pattern that supports confident de novo sequencing. Low precursor purity, co-isolation, weak signal, high charge complexity, or sparse fragmentation can leave major gaps in the ladder.
2. The peptide chemistry creates underdetermined candidates
Unknown modifications, truncation, cyclization, neutral-loss behavior, or multiple mass-shift explanations can produce outputs that look plausible but remain chemically ambiguous. Leucine/Isoleucine ambiguity is a common example of a residue call that may stay unresolved.
3. Supporting evidence does not converge
A top candidate from one spectrum is often much weaker than a consensus sequence supported by replicate agreement, repeated spectral annotation, and intact mass consistency. Software ranking can appear decisive even when the supporting evidence is thin.
4. Analyst review becomes the real bottleneck
Many teams find that the hard part is not generating candidates but defending one. When analysts need to re-annotate fragment ions, compare candidates in peptide-spectrum match comparison context, and build a validation plan from uncertain evidence, in-house analysis can slow the project more than expected.
A Method-Selection Framework for In-House Analysis vs Interpretation Support
This is a method-selection problem, not a generic troubleshooting exercise. The goal is to decide whether your current path can deliver a defensible sequence call on schedule.
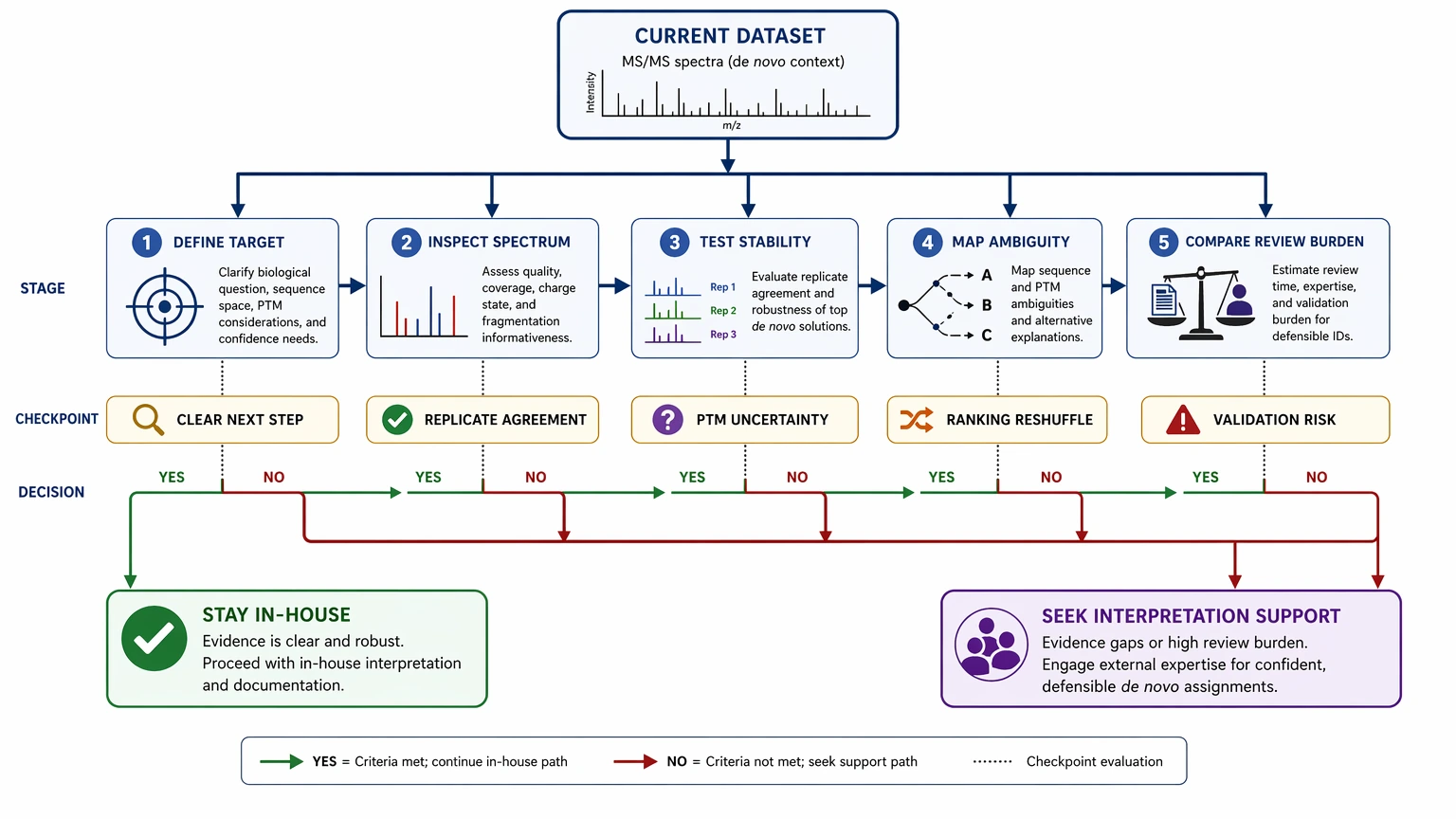
Step 1: Define what “good enough” means for this project
Separate exploratory output from report-ready output.
Ask:
A partial answer may work in early discovery. It is far less acceptable when the sequence will drive synthesis or formal technical documentation.
Step 2: Evaluate the spectrum before evaluating the software
Start with the data, not the interface or score.
| Evidence | What it supports | Main limitation | Best follow-up |
|---|---|---|---|
| Clean precursor ion isolation | Lower risk of mixed-sequence assignment | Does not ensure rich fragmentation | Inspect ladder continuity |
| Strong series of b ions and y ions | Better residue-order support | Gaps near PTMs still matter | Mark unsupported intervals |
| Replicate agreement | More stable sequence tag and ranking | Reproducibility can preserve a wrong call | Compare candidate convergence |
| High-resolution HCD | Better mass accuracy for fragment ion assignment | May underserve labile modifications | Consider ETD / EThcD if relevant |
| Diagnostic neutral losses or immonium ions | Extra support for residue or PTM reasoning | Rarely enough for a full sequence | Use as secondary evidence |
Takeaway: if the MS/MS spectrum is low-information, software tuning will not change that basic constraint.
Service Routes to Consider
For this project scenario, readers usually compare these service routes before requesting a quote or submitting samples.
Step 3: Test candidate stability, not just the top score
A defensible sequence usually behaves consistently across a few checks:
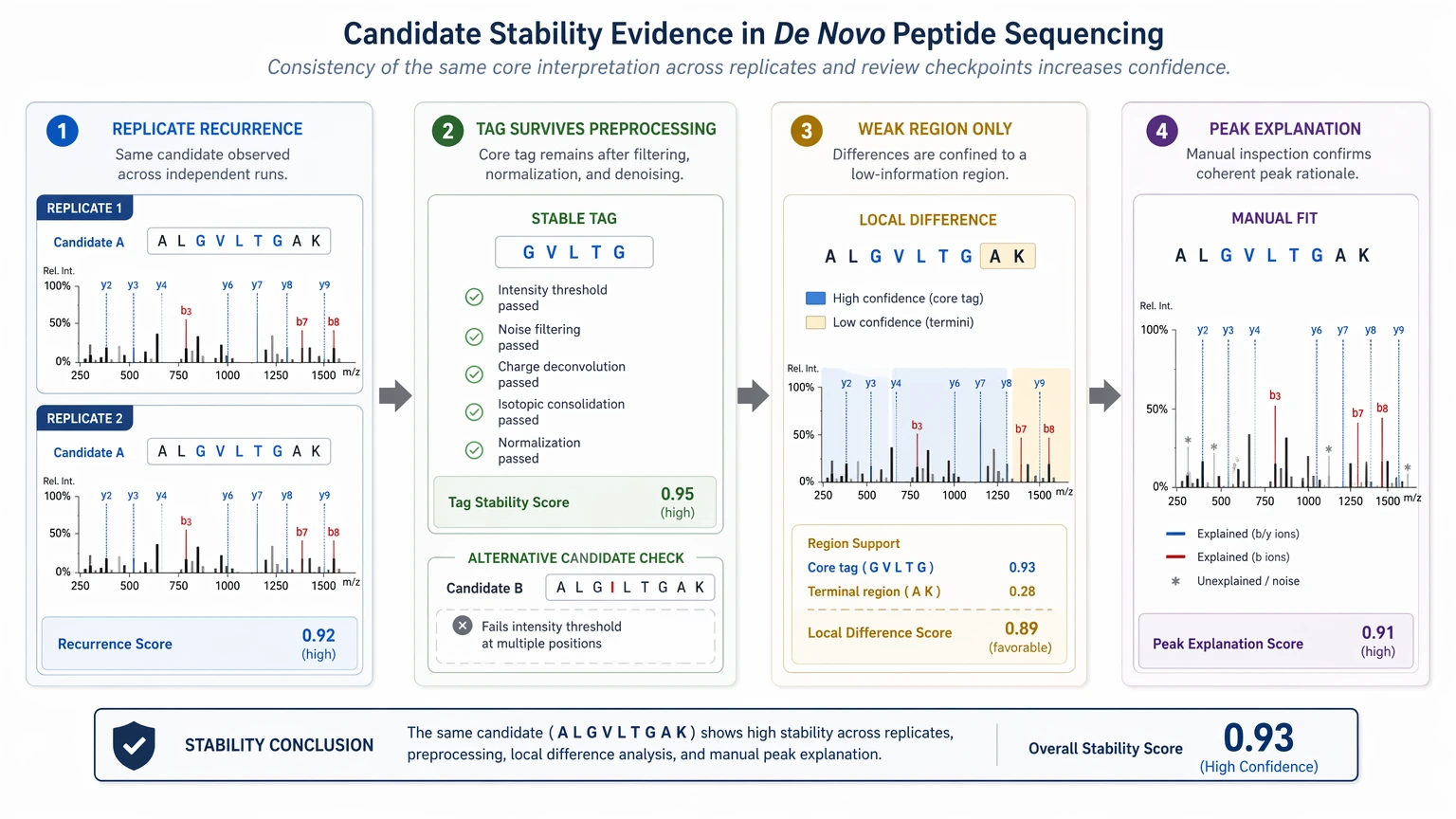
When small changes in preprocessing, PTM assumptions, or fragmentation interpretation reshuffle the ranking, sequence confidence is lower than the software output suggests.
| Scenario | Recommended workflow | Key limitation | Validation need |
|---|---|---|---|
| One dominant candidate with replicate support | Continue in-house | Terminal gaps may remain | Check intact mass consistency |
| Same sequence tag, different endings | Continue only if partial ambiguity is acceptable | Full-length call is weak | Plan synthetic peptide confirmation |
| Several unrelated top candidates | Move to interpretation support | Ranking is not converging | Cross-spectrum adjudication |
| One weak spectrum drives the call | Do not report as final | Evidence is fragile | Acquire more support |
Takeaway: stable candidate behavior matters more than a single high score.
Step 4: Map ambiguity to project risk
Not every ambiguity justifies escalation. What matters is whether the ambiguity changes the next decision.
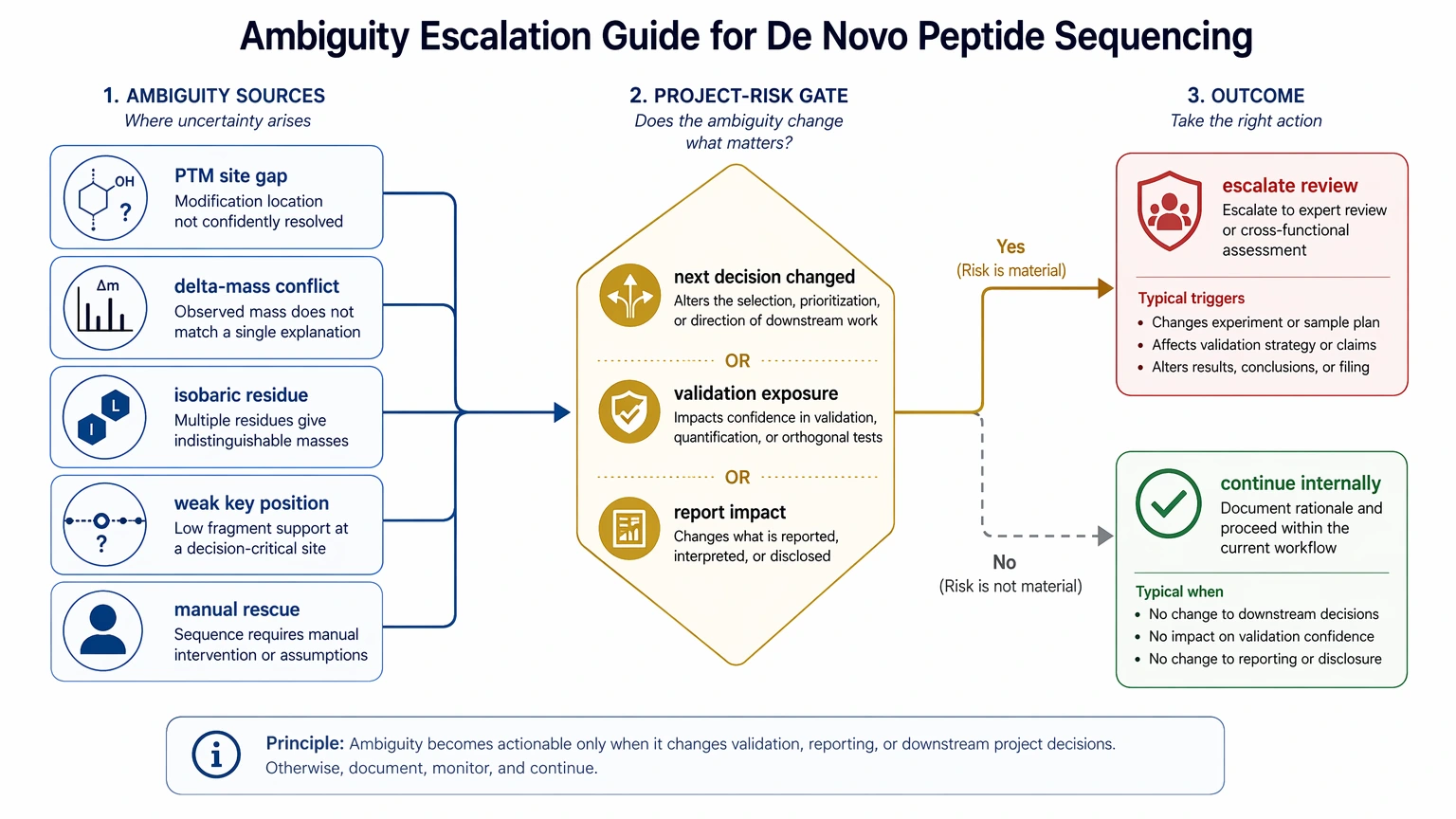
Higher-risk situations include:
This is also where fragmentation mode matters. HCD may be adequate for some unknown peptide identification tasks, while ETD / EThcD can add useful evidence for modification-rich peptides. Even so, one limit stays the same: MS/MS-based de novo sequencing may not confidently resolve every PTM site or every full-length sequence, especially when fragmentation is incomplete.
If PTM-rich or database-independent data are driving the decision, a mid-project review can save time. Teams can submit your requirements to MtoZ Biolabs for an evidence-focused assessment of spectra, fragmentation mode, and likely validation burden before spending more sample on uncertain internal iteration.
Step 5: Compare analyst time with downstream consequence
Internal analysis is often reasonable when the sample is relatively clean, sample complexity is modest, and manual review mainly confirms the software result.
External interpretation support becomes more attractive when:
That tradeoff gets sharper in impurity characterization, novel peptide discovery, and low-input projects where the first sequence decision shapes everything that follows.
Expected Results and Validation Methods
A good evaluation should produce a clearer decision, even if it does not produce a confirmed full-length sequence.
Immediate deliverables from the evaluation
You should expect:
Follow-up confirmation after the evaluation
If the sequence will guide downstream action, confirmation should be planned separately. Useful follow-up checks include:
The distinction is straightforward: the evaluation sets a defensible interpretation boundary, while confirmation tests whether that interpretation is strong enough for the next project step.
Key Cautions and Practical Limits
Before committing to one route, keep these practical limits in view.
Conclusion
The practical threshold is not whether the software can generate a sequence candidate, but whether the available LC-MS/MS evidence, candidate stability, and review burden support a sequence call you can defend. In-house analysis often fits cleaner datasets with consistent fragment ion evidence and manageable ambiguity. It fits less well when PTM reasoning, low-information spectra, or unstable rankings are driving the result.
For unknown peptide identification, impurity-related sequencing, PTM-rich targets, or other database-independent projects, the most useful outcome is a clear statement of what the data support now and what still needs confirmation. If your team needs that decision before synthesis or reporting, contact us at MtoZ Biolabs to evaluate your project around available spectra, validation goals, and the most practical level of interpretation support.
FAQ
Should we compare more than one de novo peptide sequencing software package?
Yes, if the same raw data can be processed consistently. Agreement across tools does not prove a sequence, but major disagreement is a useful warning sign that the evidence base is weak or that the scoring logic is too dependent on assumptions.
When is a sequence tag enough to move a project forward?
A sequence tag may be enough when the next step is screening, homolog searching, or planning additional acquisition. It is usually not enough when you need a full synthesis-ready sequence or a precise PTM claim.
Does external interpretation support replace internal analysts?
No. It is most useful when your internal team already understands the dataset but needs a faster or more defensible way to sort through ambiguous spectra, PTM options, or conflicting candidate lists.
What raw-data details should be captured before asking for review?
Keep the raw LC-MS/MS files, acquisition settings, fragmentation mode, precursor charge information, sample context, replicate structure, and any prior candidate annotations. Missing acquisition context can make sequence-confidence review slower and less precise.
Can database searching still contribute to a de novo project?
Yes. It can help identify contaminants, homologous regions, or expected background species. The limitation is that it cannot fully solve truly novel or modification-heavy cases where the correct answer falls outside a practical reference database.
How to order?

