





Protein Amino Acid Sequence Determination: Edman Degradation, MS Sequencing, and Method Selection
-
Edman degradation reads amino acids from the N-terminus step by step and works best for purified peptides or proteins with accessible N-termini.
-
LC-MS/MS protein sequencing digests proteins into peptides and infers sequence from accurate masses and fragment ions.
-
De novo sequencing reconstructs sequence directly from MS/MS spectra when database information is incomplete or unavailable.
-
N- and C-terminal sequencing, intact mass analysis, and peptide mapping often complement full sequence determination.
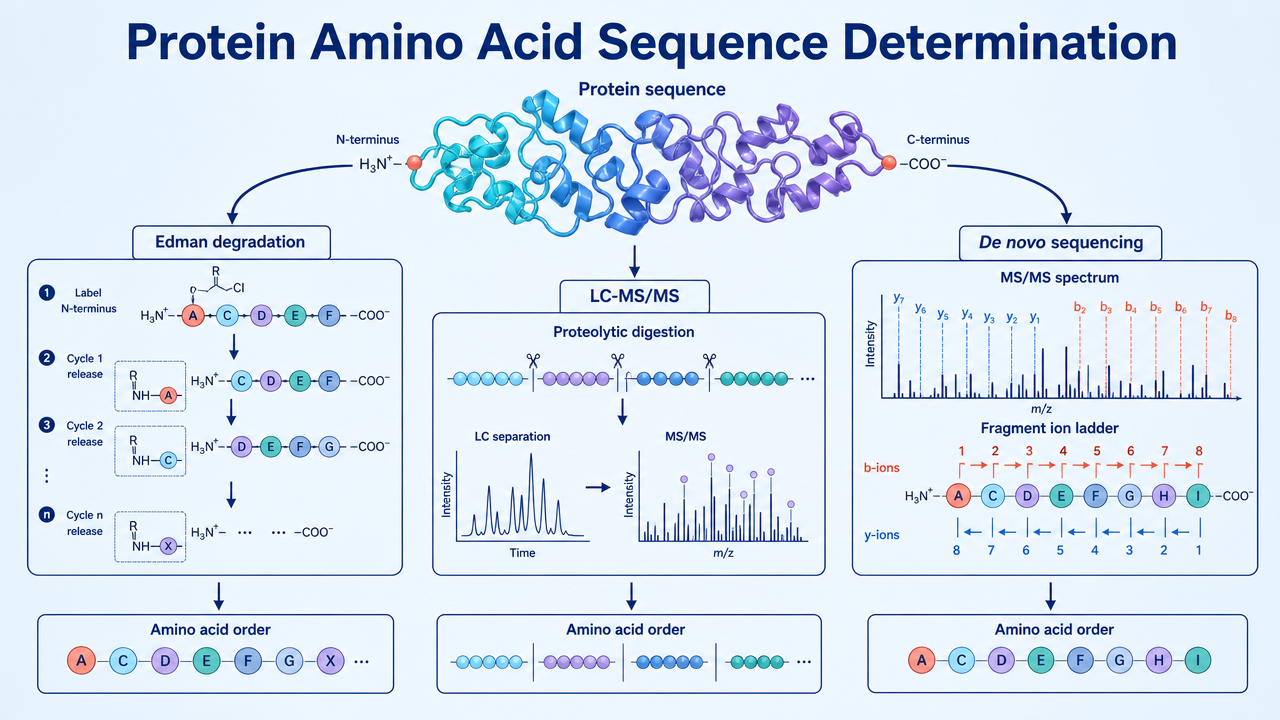
Protein amino acid sequence determination identifies the order of amino acids in a protein or peptide. The sequence can confirm protein identity, reveal mutations, support structure-function studies, guide antibody or enzyme research, and help characterize biopharmaceutical products. The two main analytical routes are Edman degradation and mass spectrometry-based sequencing; de novo MS sequencing is used when no reliable reference sequence is available.
Key Takeaways
What Does Protein Amino Acid Sequence Determination Measure?
Protein sequencing measures the linear order of amino acid residues. For short purified peptides, this may be a direct readout. For larger proteins, sequence determination usually combines multiple peptide fragments, database matching, de novo interpretation, terminal sequencing, and sometimes intact protein mass evidence.

Related Services
Protein Amino Acid Sequence Analysis Service
Mass Spectrometry-Based Protein Sequencing Service
De Novo Protein Sequencing Service
Protein De Novo Sequencing Service
Edman Based Protein Sequencing Service
Edman Degradation Protein Sequencing service
Edman Degradation
Edman degradation sequentially labels and removes amino acids from the N-terminus of a peptide or protein. It is useful when the sample is purified, the N-terminus is not blocked, and a short N-terminal sequence is needed. It can provide direct residue-by-residue information, but read length is limited and performance drops when the sample is mixed, modified, or N-terminally blocked.
Mass Spectrometry-Based Protein Sequencing
MS-based sequencing usually digests the protein into peptides, separates them by LC, and analyzes them by tandem MS. Fragment ions are matched to a reference database or interpreted to reconstruct peptide sequences. Overlapping peptides from different proteases can increase sequence coverage and help confirm ambiguous regions.
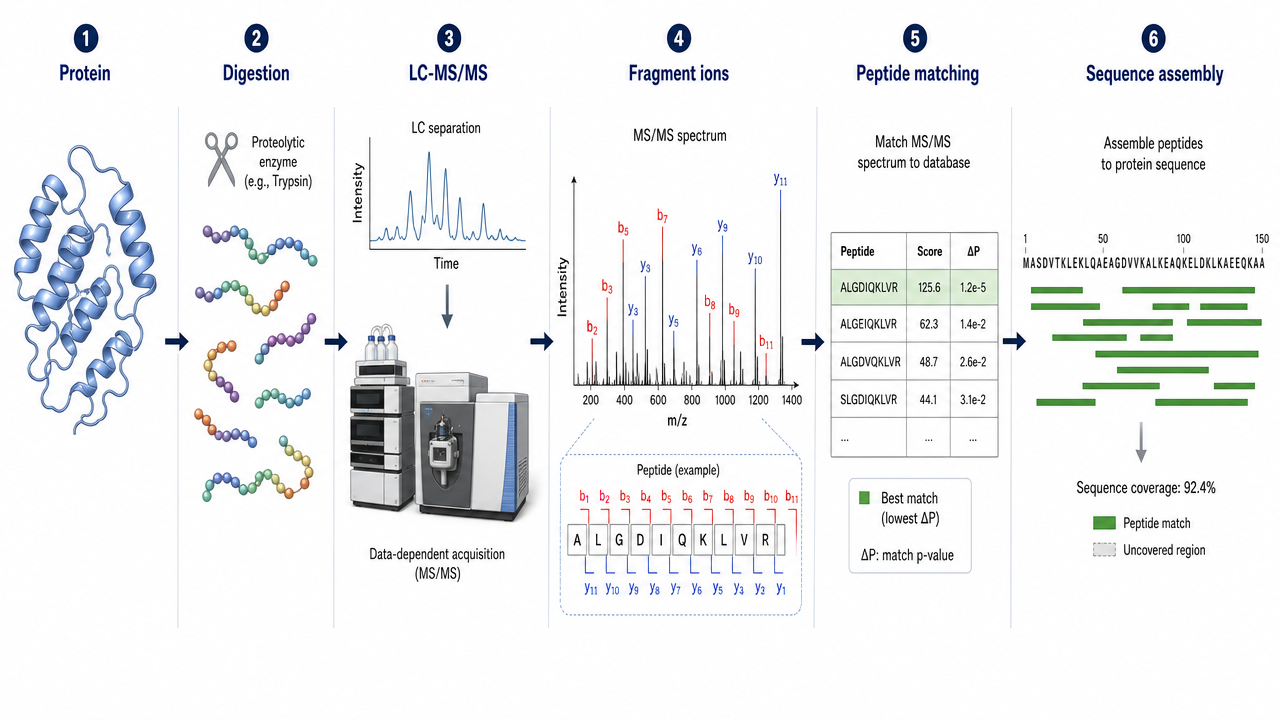
MS sequencing is sensitive and can detect modifications, mutations, terminal blocking, and unexpected sequence variants. It is usually more scalable than Edman degradation for larger proteins, but it depends on peptide coverage, spectrum quality, and data interpretation.
De Novo Sequencing
De novo sequencing reads peptide sequence from MS/MS fragment ions without relying entirely on a database. This is useful for unknown proteins, non-model organisms, antibody variable regions, unexpected mutations, or proteins where the database entry is incomplete.
The main limitation is ambiguity. Leucine and isoleucine have the same mass, some residues or modifications can be difficult to distinguish, and incomplete fragmentation can create sequence gaps.
Applications
Protein amino acid sequence determination is used for protein identity confirmation, mutation analysis, antibody characterization, enzyme engineering, species comparison, recombinant protein QC, biopharmaceutical characterization, and disease-related variant studies.
Main Limitations
No single sequencing method solves every case. Edman degradation struggles with blocked N-termini and long proteins. MS-based sequencing can miss regions that do not generate suitable peptides. De novo sequencing may produce ambiguous residues or short sequence tags when spectra are incomplete.

How to Choose a Sequencing Strategy?
| Question | Recommended Approach | Why | Main Caution |
|---|---|---|---|
| Need N-terminal residues from a purified protein? | Edman degradation | Direct stepwise N-terminal readout | Fails if N-terminus is blocked |
| Need broad sequence coverage of a protein? | LC-MS/MS peptide sequencing | Sensitive and scalable | Requires good peptide coverage |
| No reference sequence available? | De novo MS sequencing | Reconstructs sequence from spectra | Some residues remain ambiguous |
| Need terminal processing information? | N/C-terminal sequencing | Targets terminal variants | May need enrichment or tailored digestion |
FAQ
1. What is protein amino acid sequence determination?
Protein amino acid sequence determination is the process of identifying the order of amino acids in a protein or peptide using chemical sequencing, mass spectrometry, or both.
2. What is the difference between Edman degradation and MS sequencing?
Edman degradation reads residues from the N-terminus one by one. MS sequencing analyzes peptide masses and fragment ions to infer sequence, often after enzymatic digestion.
3. When is de novo protein sequencing needed?
De novo sequencing is useful when the protein is unknown, the database is incomplete, or the sample may contain unexpected variants that cannot be explained by database matching alone.
4. Can mass spectrometry detect modifications during sequencing?
Yes. LC-MS/MS can detect many modifications when the modified peptide is observed and the fragment ions support localization.
Conclusion
Protein amino acid sequence determination is strongest when the method matches the sample and the question. Edman degradation is useful for direct N-terminal reads, while LC-MS/MS provides broader and more flexible sequence evidence. For unknown proteins or incomplete databases, de novo sequencing can fill critical gaps, especially when supported by high-quality spectra and complementary workflows.
How to order?

