





De Novo Sequencing Strategy: How to Choose Read Length, Platform Mix, and Assembly Path
- Choose single-mode de novo peptide sequencing when the sample is relatively pure, the target is short or peptide-focused, and the main deliverable is a high-confidence sequence tag.
- Choose mixed fragmentation when b ions / y ions alone do not provide enough continuity, or when labile PTMs require complementary c ions / z ions evidence.
- Choose staged assembly when the target is a protein, a truncation variant, or a mixed proteoform population that needs peptide-to-protein reconstruction.
- Plan orthogonal validation early if the project cannot tolerate residue ambiguity, uncertain termini, or incomplete PTM assignment.
- precursor signals are strong, but database-supported identification is weak or contradictory
- intact mass and peptide mapping do not align cleanly
- existing spectra contain partial sequence information, but not enough for confident unknown peptide identification
- the downstream decision requires a consensus sequence, not just a list of candidate peptides
- the sample may contain more than one proteoform, making reference-driven interpretation misleading
- Peptide-level assignment: one unknown peptide or a small set of related variants
- Protein-level reconstruction: overlapping de novo tags assembled into a broader consensus sequence
- Proteoform-aware interpretation: sequence evidence linked to termini, PTMs, or intact mass context
- MS/MS spectral quality
- precursor isolation cleanliness
- continuity of the relevant fragment ion ladder
- fragment mass accuracy
- terminal coverage
- reproducibility across charge states or repeat spectra
- the sample is dominated by one peptide or a simple peptide mixture
- PTM burden is modest
- blocked termini or disulfide constraints are not a major concern
- the final deliverable does not require full protein reconstruction
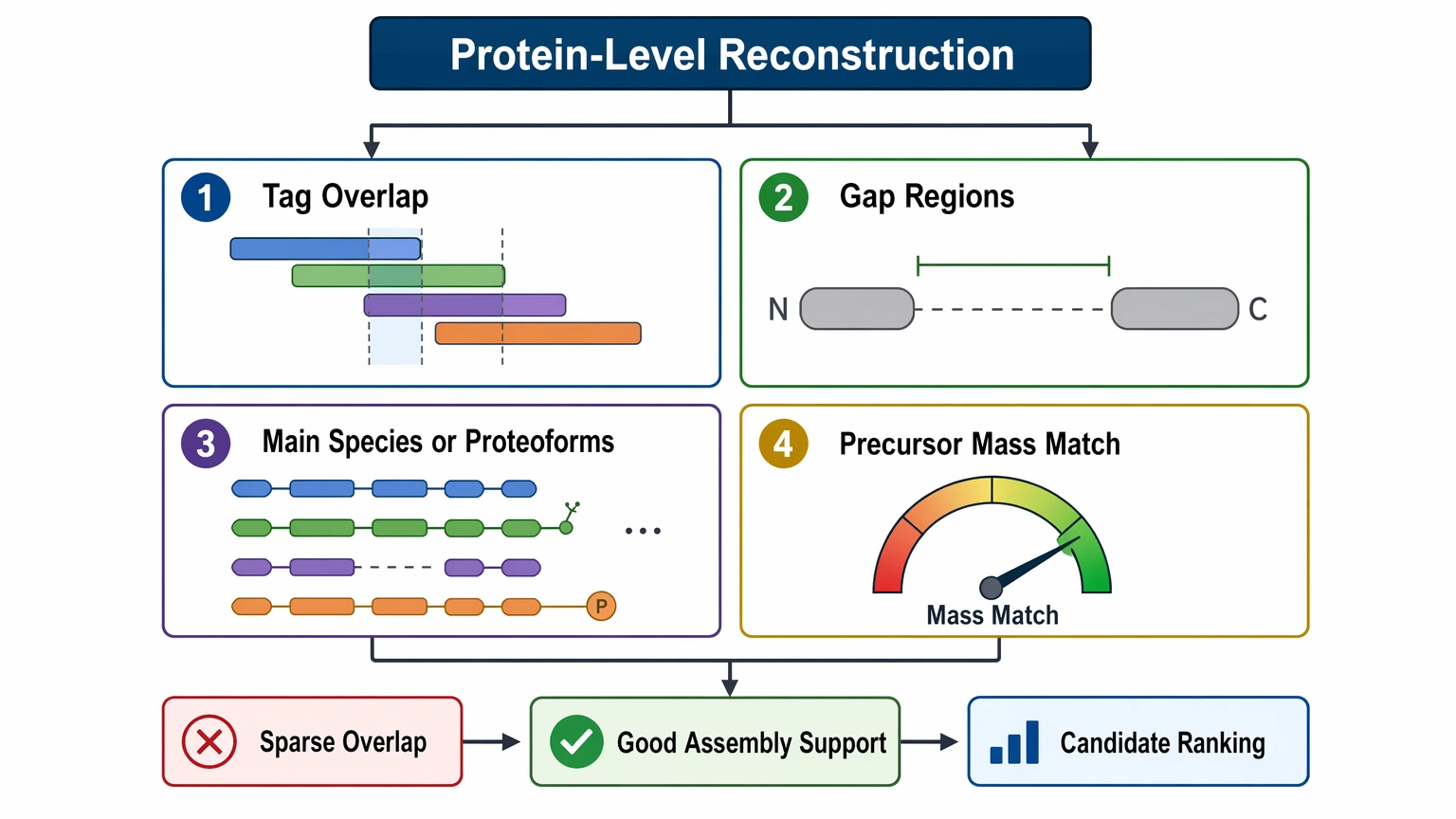
- Do the tags overlap enough to support a consensus sequence?
- Are the gaps concentrated in terminal, repetitive, or PTM-heavy regions?
- Does the sample appear to contain one main species or several related proteoforms?
- Does the proposed sequence space agree with precursor mass and intact mass measurements?
- longer and more reproducible sequence tag assignments
- clearer sequence coverage across key regions
- better agreement between proposed peptide sequences and precursor mass
- a confidence-ranked consensus sequence
- explicit annotation of unresolved residues, alternative localizations, or terminal uncertainty
- intact mass confirmation for sequence-space consistency
- targeted LC-MS/MS of high-value ambiguous regions
- peptide mapping against reconstructed candidates
- terminal checks for blocked or uncertain ends
- synthetic peptide comparison when a short candidate list remains
- Sample quality or amount limits: low abundance, detergent carryover, poor purity, or co-eluting species can shorten tags and reduce usable sequence coverage.
- Controls and repeat expectations: replicate spectra, alternate charge states, or confirmatory runs may be needed when a specific residue call carries business or scientific risk.
- Batch or contamination risk: keratin, digestion artifacts, polymer background, or carryover can create misleading candidate tags.
- Interpretation boundaries: Leu/Ile ambiguity, partial terminal coverage, and database-independent alternative candidates may remain after standard MS/MS analysis.
- When another method is the better next step: if the sample is highly heterogeneous, heavily modified, or too limited for informative fragmentation, a narrower characterization plan, additional purification, or outside support may be more productive than forcing a full de novo scope.
Choose a staged de novo sequencing strategy by starting with the kind of sequence evidence your sample is actually likely to produce. A single-fragmentation LC-MS/MS workflow is often enough for a relatively clean target with a modest modification burden. A mixed approach such as HCD plus ETD or EThcD becomes more useful when PTM localization, terminal blockage, or incomplete fragment ion ladder coverage starts to limit confidence. If the real objective is broader novel protein characterization, plan for an assembly path that connects multiple de novo peptide tags into a protein-level interpretation instead of expecting one uninterrupted read.
Quick decision guide
The main planning issue is not instrument complexity on its own. In de novo peptide sequencing and de novo protein sequencing, “read length” means how long a contiguous residue assignment remains believable based on a coherent fragment pattern, clean precursor selection, and strong fragment mass accuracy. A longer precursor does not automatically produce a longer answer.
Where de novo strategy decisions usually become necessary
This choice usually comes up after a conventional database search stops answering the real question. The sample may contain a novel sequence region, an engineered variant, a species mismatch, a truncation product, or a PTM-rich analyte that creates a peptide-spectrum match limitation. At that point, the team is no longer asking whether tandem mass spectrometry can detect the target. It is asking how much sequence confidence the available MS/MS evidence can actually support.
A few practical signs point toward de novo work:
Database-search failure by itself does not mean de novo analysis will work. The sample still needs interpretable MS/MS data, enough continuity across fragment ions, and a realistic validation path.
The main reasons de novo projects underperform
Most unsuccessful projects trace back to a small set of planning mistakes rather than a generic breakdown across the whole workflow.
1. The evidence type does not match the decision
A peptide-level project and a protein-level project do not need the same acquisition design. If the real decision depends on de novo protein sequencing, a peptide-only workflow may produce useful tags but still leave unresolved gaps between regions.
2. Fragmentation is too narrow for the sample
HCD and CID can generate strong b ions / y ions, but PTM-rich, highly charged, or structurally constrained targets may still break into short tags. In those cases, ETD or EThcD can add complementary backbone cleavage and improve PTM localization.
3. Sample heterogeneity interrupts sequence continuity
Co-isolated precursors, multiple related species, disulfide-linked regions, and partial purification can shorten tags and complicate assembly. That often shifts the likely deliverable from a single sequence claim to a confidence-ranked interpretation.
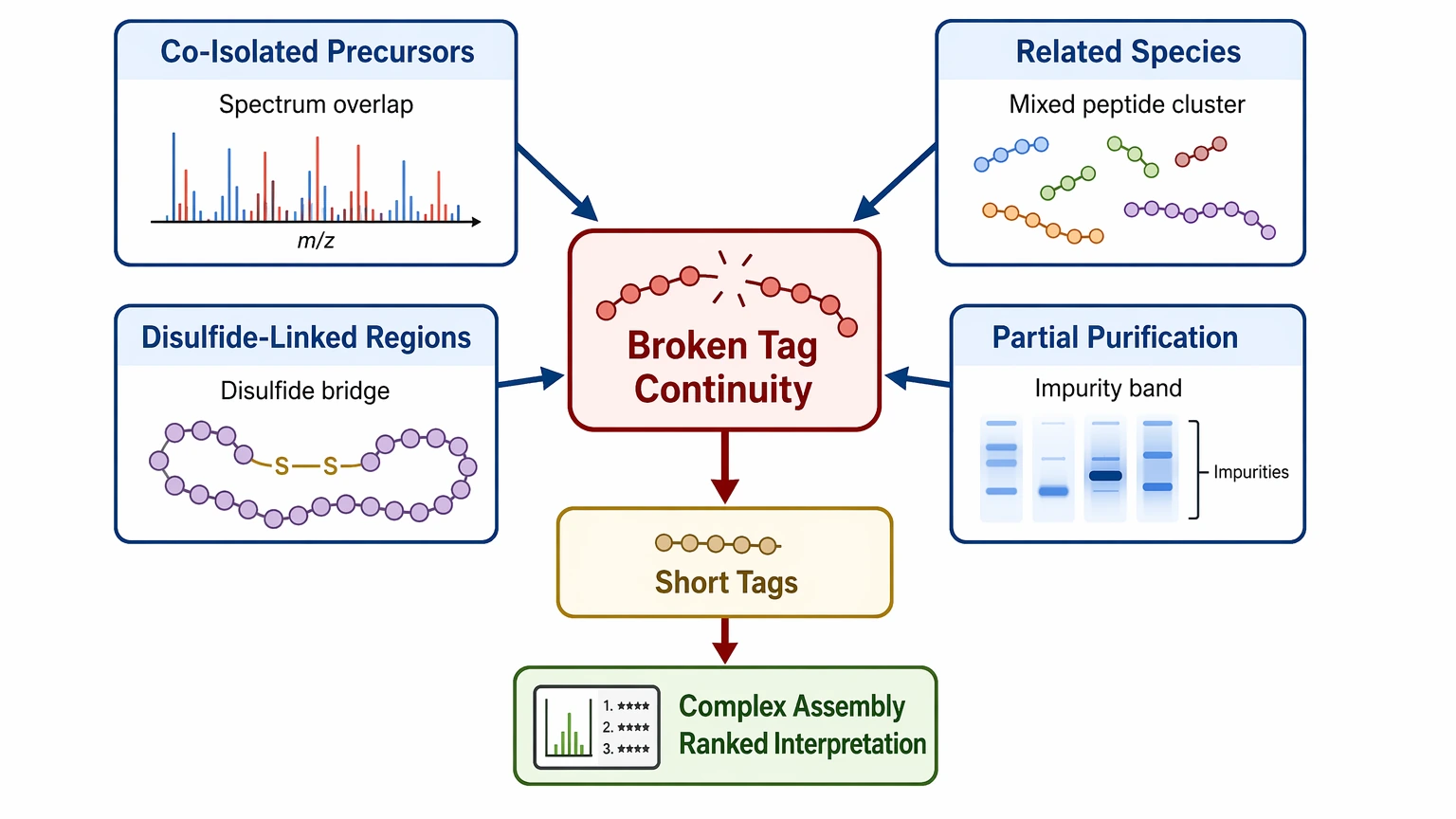
4. The team expects certainty beyond what MS/MS alone can support
Standard tandem mass spectrometry may leave Leu/Ile ambiguity, uncertain terminal residues, or more than one plausible PTM placement. That limitation should be stated directly in the project scope. Even strong LC-MS/MS evidence can still yield a confidence-ranked sequence rather than one absolute answer.
How to choose the right de novo sequencing path
This article uses a method-selection structure: first define the deliverable, then match fragmentation, then choose the assembly path, then set validation boundaries.
Step 1: Define the deliverable before you choose the workflow
Start with the decision the report must support.
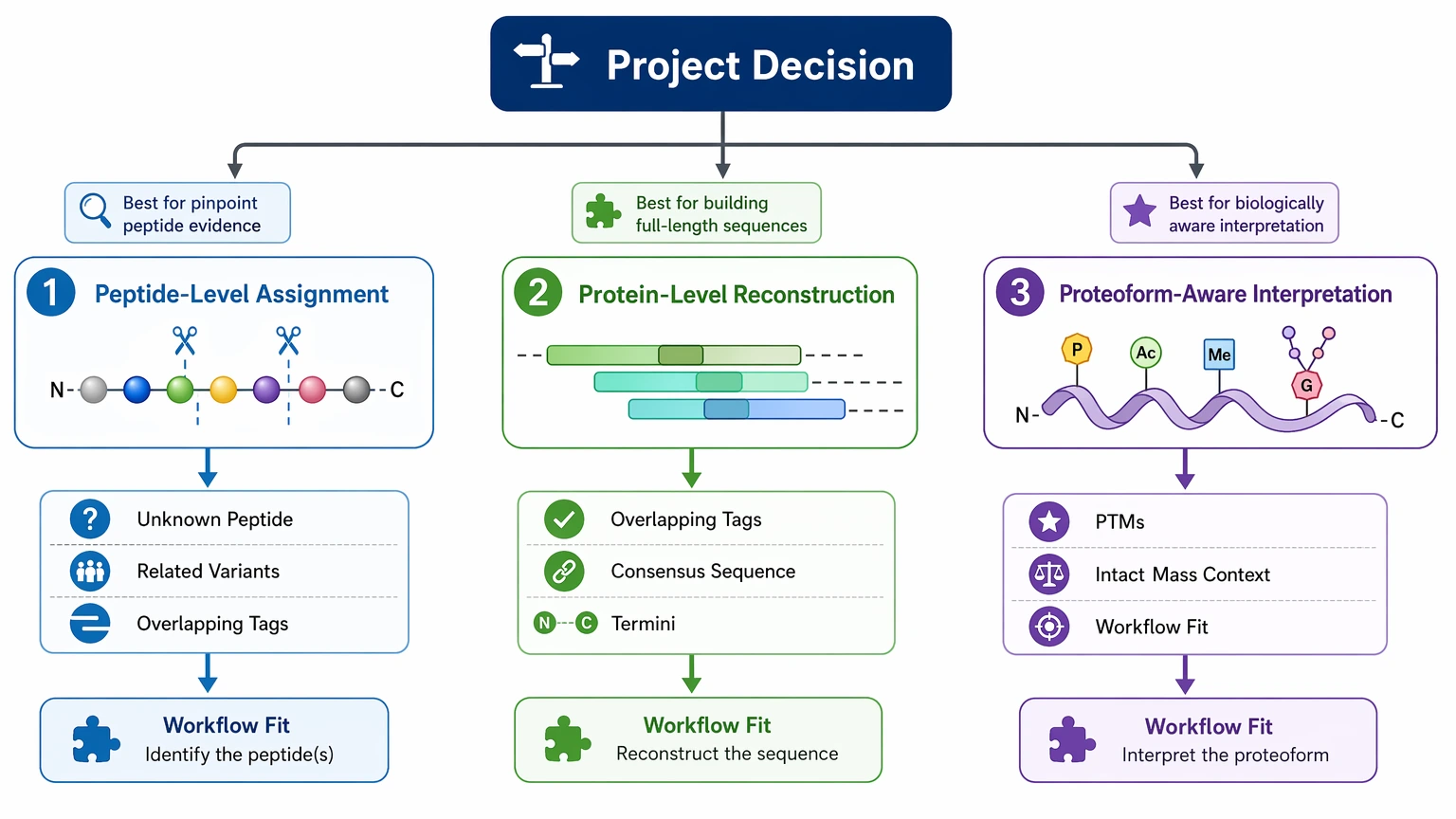
If the project only needs one peptide-level answer, a focused bottom-up design may be enough. If the question concerns truncation, engineered variation, or multiple related forms, the assembly plan matters just as much as the first run.
Step 2: Treat read length as sequence-tag continuity
For de novo MS/MS work, useful read length is the length of a supported sequence tag, not a marketing feature of the instrument. Review the evidence that actually drives sequence confidence:
The table below is a practical starting point.
| Scenario | Recommended workflow | Key limitation | Validation need |
|---|---|---|---|
| Clean unknown peptide, limited PTMs | HCD-first bottom-up de novo peptide sequencing | Terminal or isobaric uncertainty may remain | Targeted confirmation if the sequence drives a development decision |
| PTM-rich peptide with labile sites | HCD plus ETD or EThcD | Interpretation is more complex | PTM localization check plus intact mass cross-check |
| Unknown protein with partial digest evidence | Bottom-up de novo plus protein-level assembly | Gaps between peptide tags may remain | Terminal checks or targeted peptide validation |
| Mixed proteoforms or truncation variants | Staged workflow with intact mass confirmation and selective de novo runs | Overlapping species reduce tag clarity | Orthogonal confirmation is usually needed |
Takeaway: decide first whether you need a direct peptide answer or an evidence package that can support later assembly.
Service Routes to Consider
For this project scenario, readers usually compare these service routes before requesting a quote or submitting samples.
Step 3: Use a single fragmentation mode only when the sample is structurally straightforward
Single-mode acquisition is usually reasonable when the analyte population is narrow and the project can accept some residue ambiguity. HCD is often the default because it produces informative ladders for many bottom-up tasks. CID can still be useful in some settings, but the real question is whether one mode gives enough continuity across the relevant precursors.
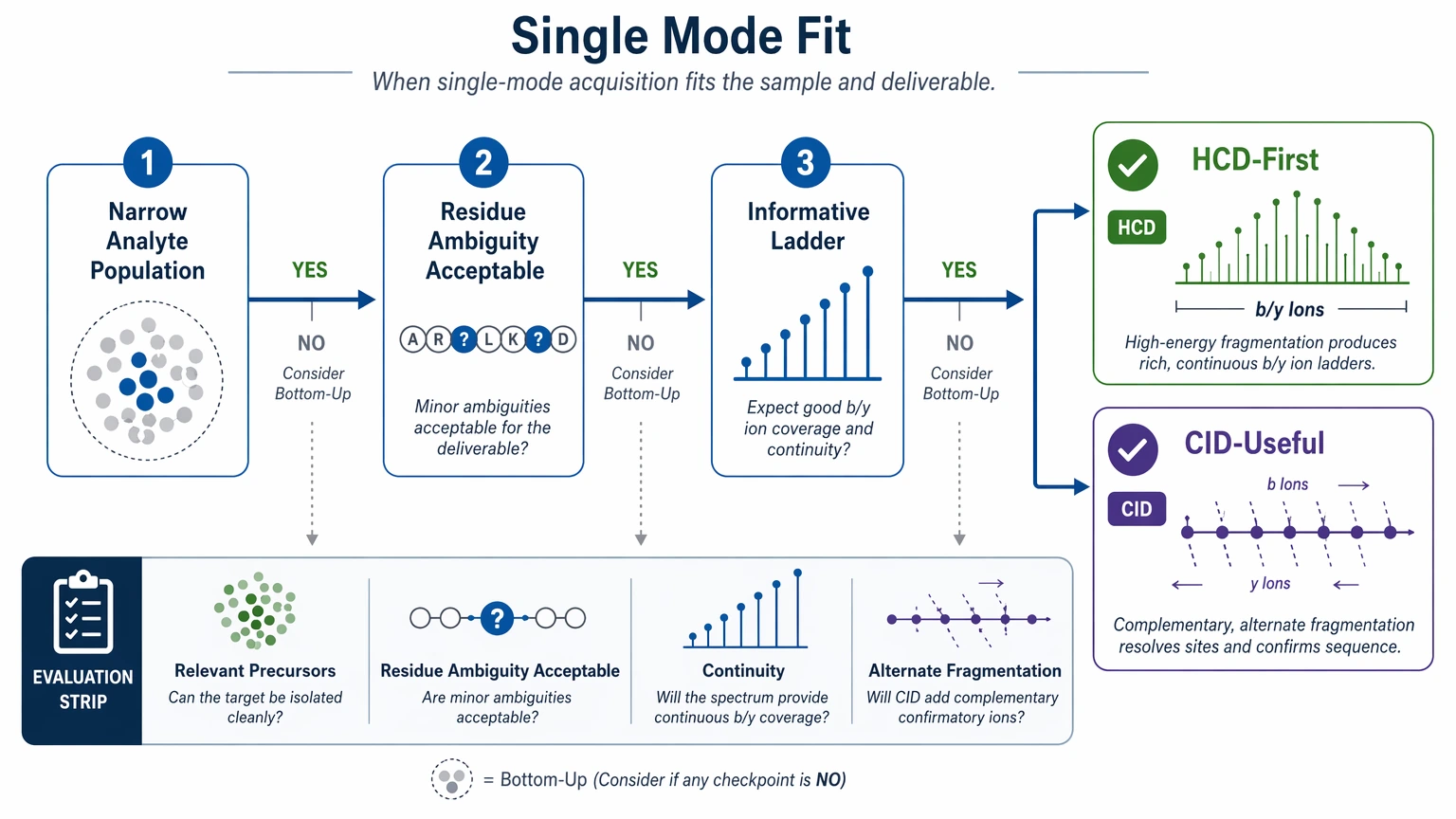
A single-fragmentation design is more defensible when:
Step 4: Add mixed fragmentation when the missing information is complementary cleavage evidence
Mixed fragmentation helps most when the problem is not lack of data volume but lack of cleavage diversity. HCD often supports b ions / y ions, while ETD and EThcD can preserve labile modifications and add c ions / z ions that clarify difficult regions.
| Evidence issue | What complementary fragmentation adds | Main limit | Follow-up |
|---|---|---|---|
| Strong HCD tags but uncertain PTM position | Better support for PTM localization | Some precursors respond poorly to ETD chemistry | Validate critical sites with targeted LC-MS/MS |
| Good precursor mass but broken sequence continuity | Nonredundant backbone evidence | Mixed samples may still fragment ambiguously | Reassess purification or fractionation |
| Poorly assigned terminal region | Extra cleavage evidence near sequence ends | Blocked termini may remain difficult | Use N- or C-terminal confirmation |
If your internal data already show incomplete ladders, PTM-heavy spectra, or disagreement between sequence tags and intact mass, this is often the point to submit your requirements for workflow review. A scoped evaluation from MtoZ Biolabs can help determine whether the better next step is mixed fragmentation, staged assembly, or an earlier sample cleanup step.
Step 5: Decide whether peptide evidence can support protein-level reconstruction
In de novo protein sequencing, protein-level interpretation is usually built from overlapping tags rather than one continuous read. Ask four questions:
When overlap is good, staged bottom-up assembly can support useful protein reconstruction. When overlap is sparse, the more honest deliverable is a ranked set of sequence candidates with explicit uncertainty markers.
Expected results and validation methods
A good strategy should improve interpretability in defined ways, not just generate more spectra.
Immediate deliverables often include:
Follow-up confirmation is separate and should be planned where it matters most. Common confirmation routes include:
A useful report separates what the LC-MS/MS data directly support from what still requires orthogonal validation.
Key cautions and practical limits
Even a well-chosen strategy has boundaries.
Conclusion
The right de novo sequencing strategy starts with the deliverable and works backward to the evidence required to support it. Single-fragmentation LC-MS/MS fits relatively clean peptide-focused projects. Mixed fragmentation becomes more useful when PTM retention, complementary cleavage, or terminal coverage limits sequence confidence. For protein-level questions, the assembly path from de novo peptide tags to a defensible consensus sequence is often the deciding factor.
This planning approach is especially useful in unknown peptide identification, impurity characterization, engineered-sequence review, and PTM-aware novel protein characterization where database search leaves major gaps. If your team needs to decide whether the likely output is a peptide-level call, a partial protein reconstruction, or a validation-guided interpretation, gather the sample context, prior spectra, and decision criteria, then contact MtoZ Biolabs to evaluate your project and discuss the most suitable sequencing and validation path before committing scarce material.
FAQ
Can de novo peptide sequencing resolve Leu and Ile directly?
Not reliably in many standard MS/MS settings. Their masses are the same, so Leu/Ile ambiguity often remains unless another line of evidence narrows the call.
When does top-down or middle-down evidence add value?
Use top-down or middle-down support when proteoform context matters, especially for terminal heterogeneity, linked modifications, or sequence regions that are hard to reconnect from peptide tags alone.
Is a longer peptide always better for de novo interpretation?
No. A longer peptide can still produce a short useful sequence tag if fragmentation is sparse or noisy. Continuity of the fragment ladder matters more than precursor length alone.
What should a procurement team ask before approving scope?
Ask what the expected deliverable is: peptide-level sequence information, partial protein reconstruction, PTM-aware interpretation, or a validation-guided candidate ranking. That question is more useful than asking for a guaranteed full sequence.
When is purification more important than adding another fragmentation mode?
Purification becomes the higher priority when co-eluting species or mixed proteoforms are already breaking precursor isolation and shortening tags. More fragmentation modes do not fix a heavily mixed precursor population.
What is the most common reporting mistake after de novo analysis?
Treating a confidence-ranked sequence interpretation as if it were full residue-level certainty. A strong report should show unresolved positions instead of hiding them.
How to order?

