





De Novo Sequencing by ESI-MS/MS: Troubleshooting Fragment Coverage and Sequence Ambiguity
- discontinuous b ions or y ions
- strong peaks that do not support one residue order
- a recurring gap near one terminus
- neutral loss or internal fragment patterns that dominate the spectrum
- different top candidates across technical reruns
- unresolved isobaric residues at a project-critical position
- whether the precursor ion isotope envelope supports the assigned monoisotopic peak
- whether neighboring peaks in the isolation window could contribute product ions
- whether two partial ladders diverge after a shared start
- whether unexplained peaks form a second mass family across reruns
- a provisional sequence tag that anchors the central region
- a constrained candidate set with explicit gap positions
- a confirmed final sequence supported by follow-up evidence
- a longer or cleaner sequence tag
- a reduced set of plausible residue assignments
- a defined ambiguous region rather than diffuse uncertainty
- a decision on whether the current precursor ion is worth rerunning
- targeted repeat acquisition of the same precursor ion under cleaner isolation
- comparison of technical reruns for fragment continuity and mass error consistency
- intact mass checks against the retained candidate family
- orthogonal validation for unresolved isobaric residues
- alternative digestion or termini-focused analysis for protein-level reconstruction
A weak de novo sequencing by ESI-MS/MS result usually becomes interpretable only after you separate a spectrum-quality issue from a real sequence-confidence limit. The fastest useful checks are precursor isolation cleanliness, charge state suitability, continuity of b ions and y ions, and whether an unexplained mass shift fits a post-translational modification (PTM) better than a new residue path.
For unknown peptide identification, repeating the same LC-MS/MS settings is rarely the smartest first move. A partial sequence tag can still be useful, but only when the assigned product ion pattern stays coherent, the mass error remains internally consistent, and the ambiguous positions are reported honestly instead of being forced into a complete call. One limitation is worth stating at the start: routine tandem mass spectrometry often cannot resolve every sequence ambiguity from a single spectrum, especially when isobaric residues, PTMs, or database search limitation leave more than one plausible interpretation.
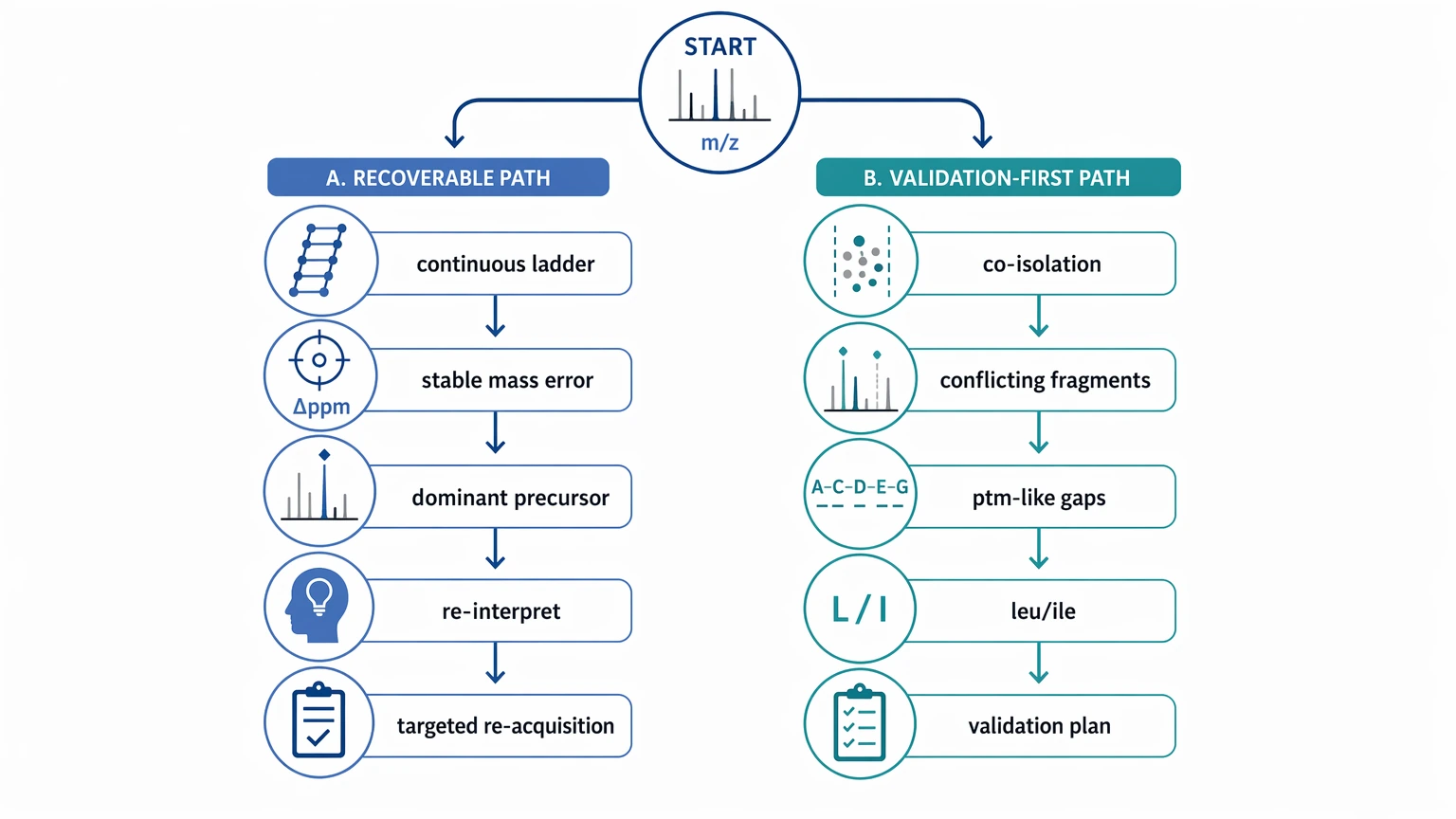
Quick Decision Check
If your spectrum shows a partly continuous ion ladder, stable mass error, and one dominant precursor ion, start with re-interpretation and targeted re-acquisition.
If it shows co-isolation, conflicting fragment families, repeated PTM-like mass gaps, or irreducible Leucine/Isoleucine ambiguity, move early to a broader validation plan instead of forcing a full sequence call.
Where This Failure State Usually Appears
This issue usually shows up after de novo sequencing software returns a short sequence tag, several close-scoring candidates, or residue assignment gaps that do not fit the precursor ion mass or sample context. Common warning signs include:
These symptoms matter because they change downstream decisions. You may need to decide whether an impurity is worth tracking, whether a modified peptide is real, whether a truncation product is present, or whether peptide-level evidence can support a larger de novo sequencing effort. In that setting, sequence confidence matters more than producing a longer-looking answer.
The Four Cause Categories That Usually Matter Most
For this troubleshooting scenario, four cause categories account for most low-confidence outcomes.
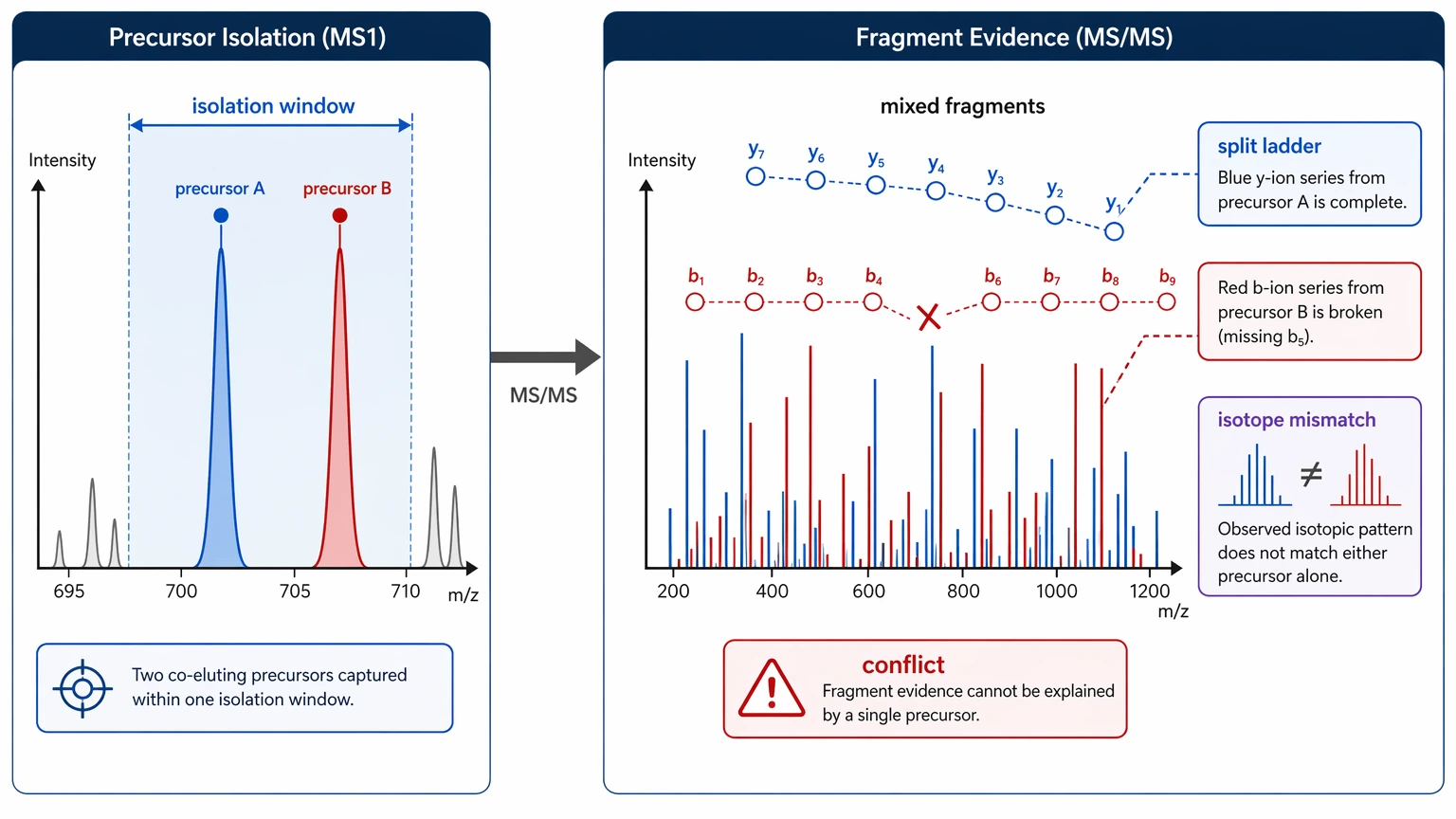
1. Poor precursor isolation creates a chimeric spectrum
If the isolation window captures more than one precursor ion, the resulting product ion population may represent multiple peptides. That creates incompatible mass-difference paths, branched sequence tags, and residue assignment conflicts that no single sequence can explain.
2. The selected charge state does not fragment into a useful ladder
A precursor ion may be abundant and still fragment poorly for de novo sequencing. You may see intense peaks, yet too few sequence-informative product ions to connect the peptide backbone across the full region of interest.
3. PTMs or unexpected termini distort the expected mass logic
A PTM, cyclization event, truncation, blocked terminus, adduct, or derivatization artifact can shift local mass differences enough to mislead automated scoring. In those cases, the software often prefers a convenient but incorrect residue assignment.
4. The ambiguity is fundamental to the evidence
Some sequence ambiguity remains even in otherwise good spectra. Leucine/Isoleucine ambiguity is the classic example, but similar uncertainty can appear whenever fragment coverage does not cross the decisive position or the evidence supports more than one chemically plausible path.
A Troubleshooting Path for Recoverable vs. Non-Recoverable Results
This article uses a troubleshooting and data-interpretation structure rather than a generic LC-MS/MS checklist. The goal is to decide what still deserves confidence, what needs a rerun, and what should be validated elsewhere.
Step 1: Classify the failure pattern before changing settings
Start by deciding whether the problem is sparse coverage, mixed fragmentation, a modification-driven gap, or a true residue-level limit.
| Scenario | Recommended workflow | Key limitation | Validation need |
|---|---|---|---|
| Short but coherent sequence tag | Reinspect product ion assignments and charge state | Terminal region may stay open | Targeted rerun |
| Multiple plausible candidates with crowded peaks | Check precursor isolation and isotope envelope | Chimeric spectrum risk | Cleaner isolation or fraction |
| Repeated fixed mass offset in one region | Test PTM or terminus hypotheses | Software may overfit residues | Intact mass context |
| Strong spectrum with unresolved Leucine/Isoleucine ambiguity | Keep the site constrained, not overcalled | Routine ESI-MS/MS may not distinguish it | Orthogonal confirmation |
Takeaway: the first decision is not “How do I get a full sequence?” but “What kind of failure am I actually looking at?”
Step 2: Exclude chimeric spectrum behavior early
A chimeric spectrum is one of the quickest ways to lose both time and sample. Look for fragment clusters that suggest more than one backbone, isotope-envelope irregularities around the selected precursor ion, or local ion ladders that cannot be connected without contradiction.
Service Routes to Consider
For this project scenario, readers usually compare these service routes before requesting a quote or submitting samples.
Useful checks include:
For de novo sequencing, a moderate-intensity but clean MS/MS event is often worth more than a high-intensity mixed spectrum.
Step 3: Ask whether the fragmentation pattern is sequence-informative
For de novo sequencing, peak count by itself is not enough. What matters is whether the product ion pattern produces a defensible sequence tag across the decision-critical region.
| Evidence | What it supports | Limitation | Follow-up |
|---|---|---|---|
| Consecutive central y ions or b ions | Actionable local sequence tag | Ends may remain uncertain | Keep as provisional anchor |
| Neutral loss dominates | Poor sequence-informative fragmentation | More signal may not help | Adjust charge state or fragmentation |
| Internal fragments outnumber terminal ions | Overfragmentation or complex gas-phase behavior | Residue order remains weak | Re-acquire under changed conditions |
| Same gap across reruns | Possible sequence-intrinsic or PTM issue | Repetition alone adds little | Expand validation plan |
If one charge state gives you cleaner continuity than another, follow that evidence even when the less informative precursor ion is more abundant.
Step 4: Test chemistry before accepting a residue assignment
When one region fails repeatedly, ask whether the problem is chemical rather than sequence-based. Review intact mass consistency, recurring neutral loss, immonium ion support, and whether the unexplained shift is localized to one terminus or one labile region.
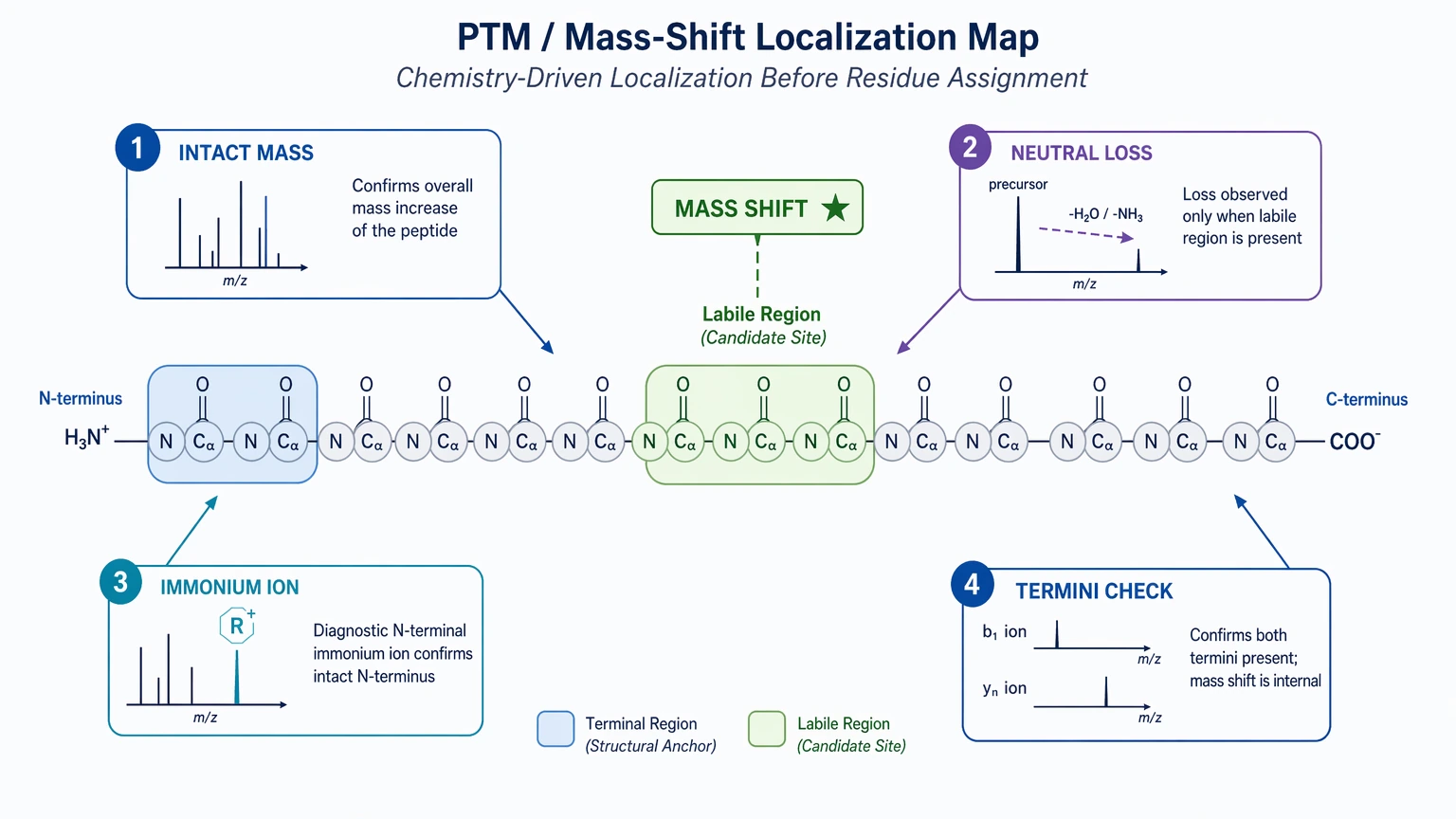
This step matters even more when database search limitation leaves little external context. For novel, modified, or non-reference peptides, relaxed scoring thresholds can create false confidence. A more defensible approach is to narrow the hypothesis space with PTM-aware interpretation.
Step 5: Separate provisional sequence information from a final call
A useful de novo result does not always mean a complete sequence. In practice, you may end up with three different deliverables:
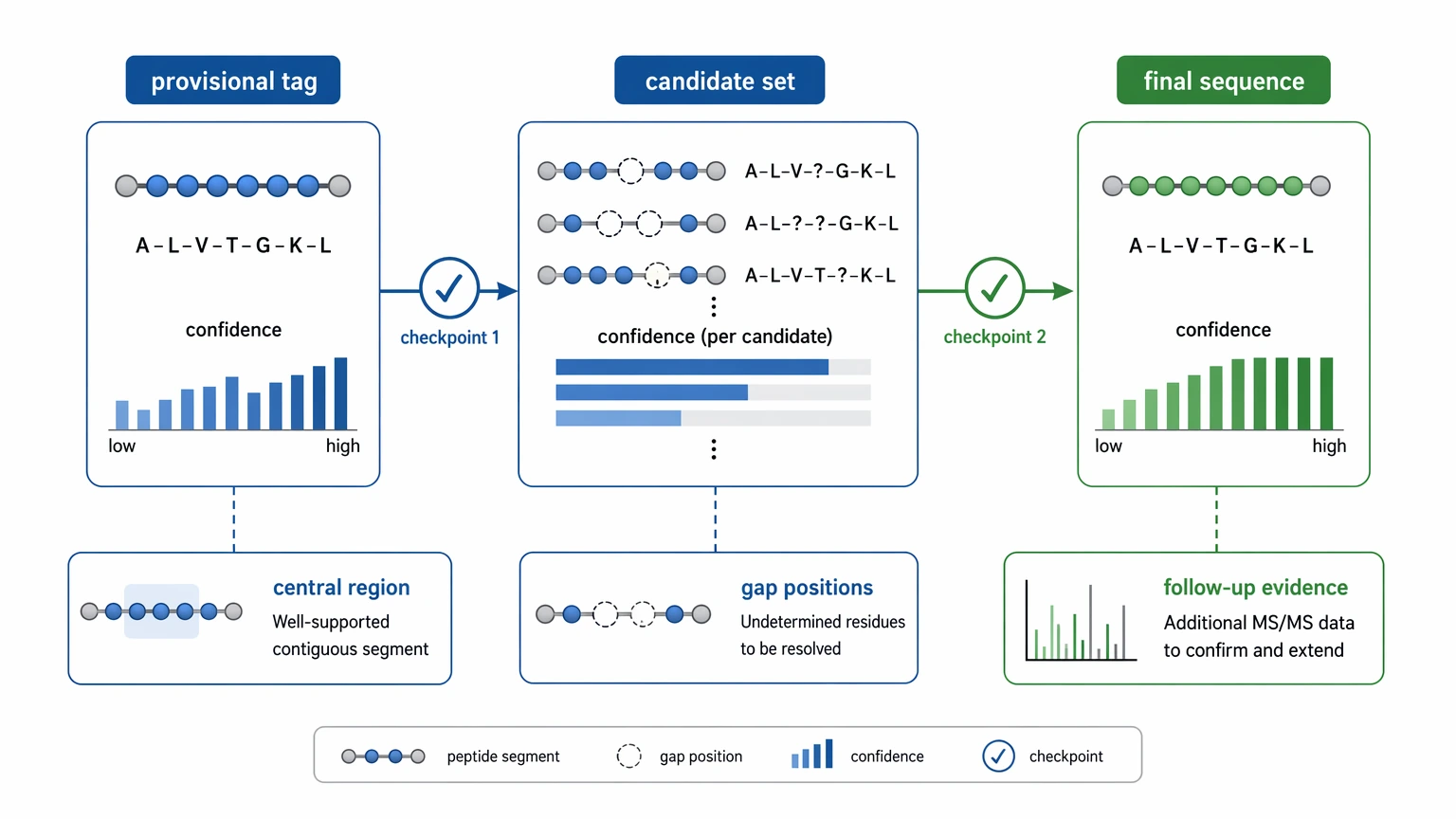
That distinction protects sequence confidence. If a critical site depends on one weak fragment, or if competing candidates require a similar number of assumptions, the result should remain provisional. Clear uncertainty is better than filling the gap with an unsupported residue assignment.
If you need an external review of whether the current spectra justify another internal rerun or a redesigned de novo sequencing workflow, you can submit your requirements to MtoZ Biolabs and evaluate your project against available De Novo Peptide Sequencing Services, De Novo Sequencing Mass Spectrometry Service, or the LC-MS/MS Analytical Service.
Expected Results and Validation Methods
A good troubleshooting pass should leave you with a clearer decision state, not a forced promise of complete recovery.
Immediate deliverables
The immediate output is usually one of the following:
These are interpretation deliverables. They show what can be trusted now.
Follow-up confirmation
Confirmation should match the type of uncertainty. Suitable follow-up steps include:
Leucine/Isoleucine ambiguity deserves an explicit note here: if routine ESI-MS/MS evidence does not distinguish the site, follow-up confirmation may narrow the call, but the original spectrum alone should not be treated as definitive.
Key Cautions and Practical Limits
Even good troubleshooting logic has boundaries.
Sample quality or amount limits
Low-abundance unknowns, heterogeneous fractions, and limited sample mass can produce interpretable local evidence without supporting a stable full-sequence call. If additional injections would likely consume the remaining material without changing the main bottleneck, stop and redesign.
Controls and repeat expectations
A rerun should test a specific question, such as narrower precursor isolation or a more informative charge state. Repeats without a hypothesis rarely improve sequence confidence.
Batch and contamination risk
Carryover, mixed digest products, or partially overlapping fractions can add product ions that imitate alternate sequence logic. Review blanks, adjacent injections, and fraction purity when the spectrum looks unexpectedly crowded.
Interpretation boundaries
Sequence tag, constrained candidate set, and confirmed final sequence are not interchangeable outputs. PTMs, database search limitation, and incomplete fragment coverage can leave uncertainty that is real rather than fixable.
When another method or outside support is the better next step
If the same ambiguous region persists across reruns, if precursor isolation remains mixed, or if peptide-level tags do not assemble into a larger protein context, another method may be more efficient than continued internal troubleshooting. In that situation, contact MtoZ Biolabs to submit your requirements, discuss the sample and data package, and evaluate your project before more material is spent.
Conclusion
De novo sequencing by ESI-MS/MS becomes more manageable when you classify the failure mode before changing acquisition settings. Clean precursor isolation, sequence-informative b ions and y ions, stable mass error, and careful treatment of PTM-related shifts and isobaric residues are the checkpoints that usually decide whether a result is still recoverable. This approach fits unknown peptide identification, impurity assessment, modified peptide review, and early protein-level sequencing triage. If your current data package still contains coherent sequence tags but unresolved decision points, prepare the raw spectra, precursor ion list, intact mass context, suspected PTMs, and your validation plan before seeking consultation.
FAQ
When is a short sequence tag still useful?
A short tag is useful when it spans the region that drives the project decision, such as an impurity motif, cleavage site, or modified segment. It does not need to cover the entire peptide to guide the next experiment.
What makes a spectrum unsuitable for de novo sequencing even if the signal is strong?
Strong signal is not enough if the product ion pattern is internally inconsistent. Crowded peaks, parallel fragment families, or dominant neutral loss can make a spectrum less interpretable than a weaker but cleaner MS/MS event.
Can intact mass help when the MS/MS sequence is incomplete?
Yes. Intact mass can rule out candidate families that do not fit the measured precursor context, especially when the main uncertainty is a PTM, truncation, or blocked terminus rather than residue order alone.
Should I trust the top de novo software score if replicate runs disagree?
Not by itself. If technical reruns change the residue assignment rather than just the peak intensity, the score is probably reflecting unstable evidence rather than stable sequence confidence.
What project details make an external review faster?
The most useful package includes raw files, acquisition settings, precursor ion m/z and charge state, intact mass data, suspected PTMs, sample origin, purification history, and a clear statement of which residue assignment or sequence ambiguity affects the project decision.
How to order?

