





De Novo Peptide Sequencing Algorithm Explained: How Spectrum Evidence Becomes Sequence Confidence
- A de novo peptide sequencing algorithm is most convincing when the candidate sequence shows continuous b ions or y ions, strong precursor mass consistency, limited unexplained peaks, and clear rank separation from other candidates.
- It is less convincing at specific positions when the spectrum contains missing-ion gaps, co-fragmentation, PTM-related mass shifts, or leucine/isoleucine ambiguity.
- A high sequence-level confidence does not mean every residue is equally secure.
- A reported sequence is often sufficient to guide validation even when one or two positions remain unresolved.
- One explicit limit remains: standard MS/MS data often cannot distinguish all isobaric residues or fully localize every PTM, so sequence confidence is not the same thing as complete structural certainty.
- Sample quality or amount limits: low signal, poor isolation purity, or insufficient material can reduce ladder continuity and shorten usable sequence tags.
- Controls and repeat expectations: spectra with mixed evidence often justify repeat acquisition, cleaner precursor isolation, or replicate confirmation before sequence-based decisions.
- Batch and contamination risk: co-isolation, carryover, and mixed modification states can introduce fragment ions that the algorithm may incorrectly fold into a candidate path.
- Interpretation boundaries: a candidate sequence may support motif discovery, synthesis planning, or targeted validation without supporting a full claim of residue-resolved certainty across the entire peptide.
- When another method is the better next step: if the decision depends on exact leucine/isoleucine discrimination, full PTM localization, or protein-level architecture beyond peptide evidence, a complementary method or outside support is often more informative than repeating the same de novo call.
A de novo peptide sequencing algorithm matters when database search cannot account for an unknown peptide, but its output is better read as graded spectral evidence than as a final statement of residue certainty. The practical issue is not whether the software prints a confidence score. It is whether specific residues are directly backed by fragment ions, which positions are inferred across gaps, and whether the remaining uncertainty is acceptable for the next experiment.
That distinction becomes important in novelty-driven LC-MS/MS work. When the analyte comes from a non-model organism, an engineered peptide region, an unexpected variant, or a PTM-rich sample, the algorithm has to rebuild amino acid order from the fragmentation spectrum itself. Teams comparing a workflow, vendor, or validation plan therefore need to know how spectral evidence turns into a candidate sequence, and where that logic starts to weaken.
Quick Decision Guide
What the Algorithm Is Solving
De novo peptide sequencing infers amino acid order directly from tandem mass spectrometry (MS/MS) data without requiring an exact database match. Instead of asking whether a measured spectrum fits a known peptide, the de novo sequencing algorithm asks which candidate sequence best explains the observed fragment ion pattern under a precursor mass constraint.
That difference is central in discovery work. A database search performs well when the sequence space is already represented. It loses power when the peptide is absent from the reference set, truncated in an unexpected way, heavily modified, or derived from a poorly annotated source. In those settings, de novo peptide sequencing does not replace database methods outright, but it can provide sequence tags, partial reconstructions, or ranked candidate sequences that a database-first workflow may miss.
How a De Novo Sequencing Algorithm Builds Candidate Sequences
Most workflows follow the same basic evidence path, even though scoring functions differ across software packages.
Spectrum preprocessing and peak list construction
The algorithm starts with an LC-MS/MS fragmentation spectrum linked to a precursor ion. It converts the raw signal into a peak list, removes obvious noise, interprets isotopes and charge states where possible, and applies mass calibration or intensity normalization if the workflow includes those steps. This stage matters because later graph construction assumes the retained peaks mainly reflect chemically meaningful fragment ions rather than random signal.
Fragment-ion interpretation
Next, the algorithm tests whether mass differences between peaks match plausible residue masses. In bottom-up de novo peptide sequencing, b ions and y ions usually provide the main backbone evidence, although other fragment ion types, neutral losses, and charge variants may also contribute under certain fragmentation conditions. Better mass accuracy lowers the number of accidental matches and helps the algorithm separate real residue steps from coincidental mass differences.
Graph construction from mass differences
Many de novo sequencing algorithms represent the fragmentation spectrum as a mass difference graph. Peaks become nodes, and edges connect nodes whose mass difference corresponds to one residue or a small residue combination. The precursor mass constraint and terminal masses help anchor possible start and end points. At this stage, the search space can expand quickly if the spectrum is noisy or if multiple explanations fit the same mass gap.
Candidate generation and path scoring
The algorithm then searches for the highest-scoring path through that graph. Path scoring usually rewards ion-series continuity, agreement with high-intensity assigned peaks, precursor mass consistency, and plausible fragmentation behavior. It also penalizes unexplained peaks, unsupported jumps, or paths that rely too heavily on missing-ion tolerance. Some tools use machine-learned scoring models, but the basic logic is still the same: accumulate evidence across the peptide backbone.
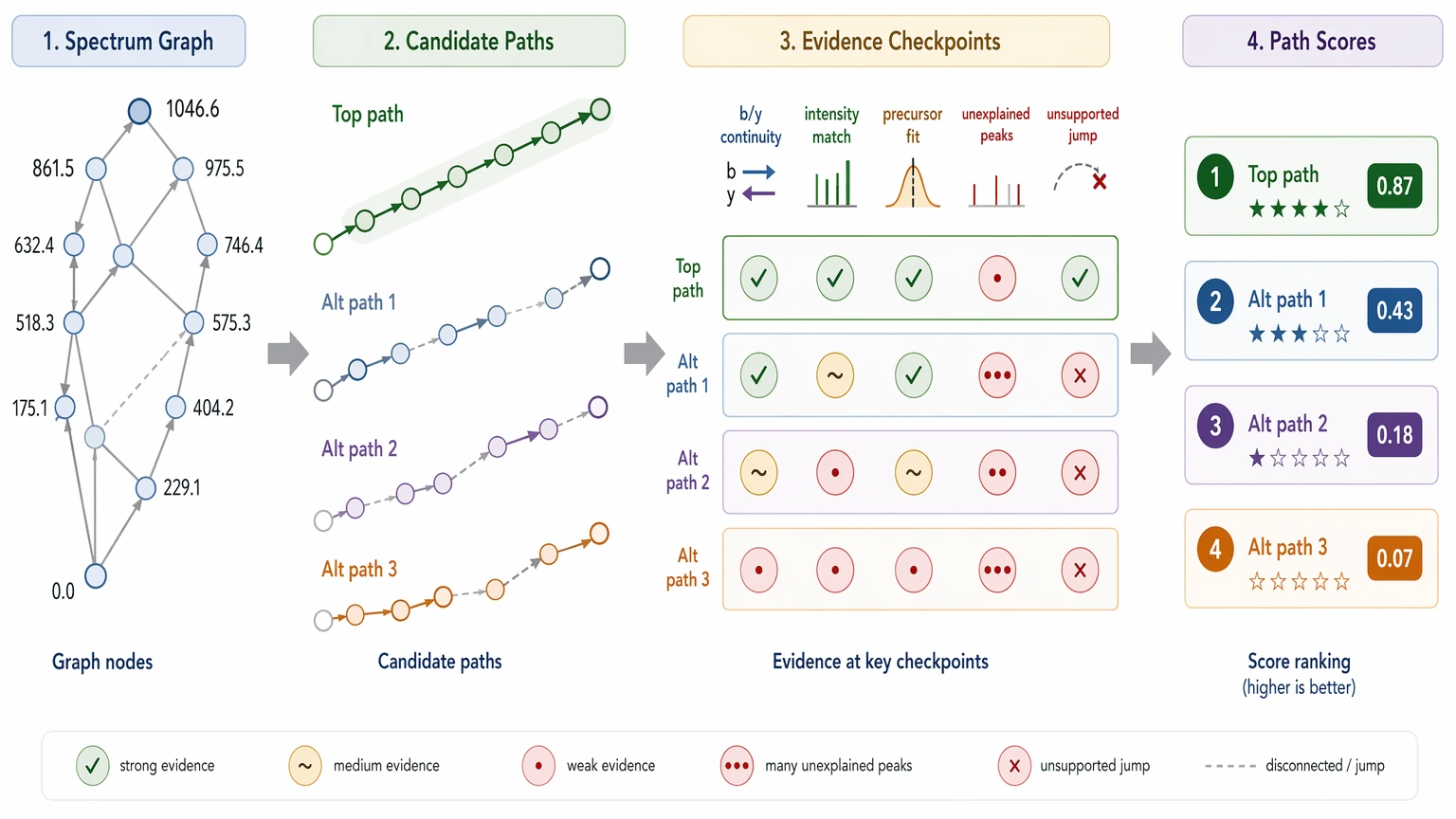
How Spectrum Evidence Becomes Confidence
Confidence in de novo peptide sequencing comes from several layers of support, not from a single binary rule.
A residue gains residue-level confidence when adjacent fragment ions define its mass difference directly. The strongest case usually appears when both N-terminal and C-terminal evidence meet at the same position, producing a coherent local b-ion and y-ion ladder. Confidence drops when a residue sits inside a gap, when the assignment depends mostly on mass balance, or when more than one amino acid explanation still fits.
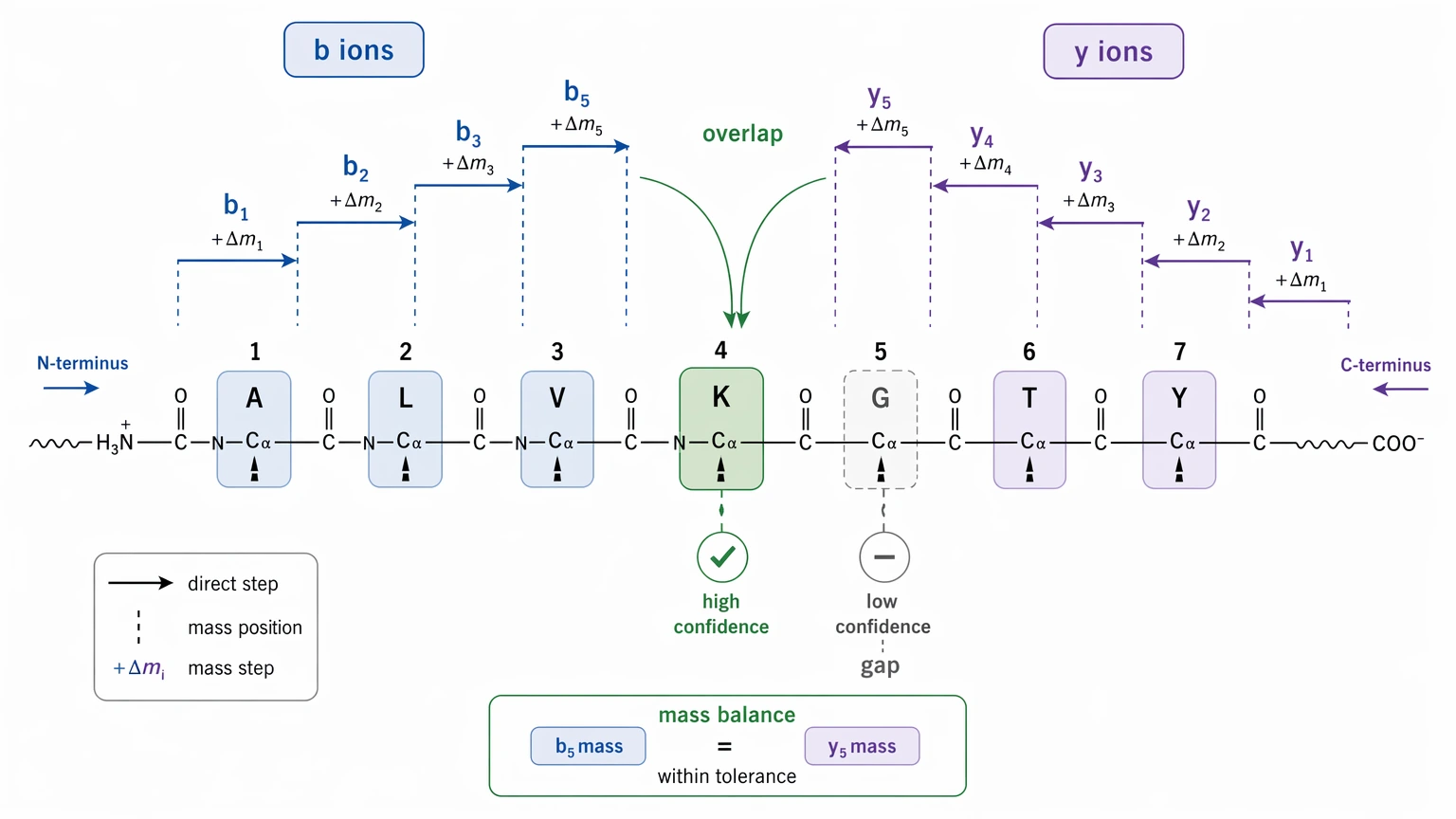
At the full-sequence level, sequence-level confidence reflects how well the candidate sequence explains the fragmentation spectrum as a whole. That includes precursor mass consistency, backbone coverage, the number and distribution of unexplained peaks, intensity support for assigned fragment ions, and rank separation from competing candidates.
The table below summarizes how common evidence types should be read.
| Evidence | What it supports | Main limitation | Best next check |
|---|---|---|---|
| Continuous b ions | N-terminal residue order across linked positions | Missing internal steps weaken local certainty | Inspect the gap boundaries |
| Continuous y ions | C-terminal residue order across linked positions | May leave the opposite terminus weak | Compare overlap with b ions |
| Overlapping b/y support | Strong residue-level confidence | Does not resolve all isobaric residues | Check ambiguity notes |
| Precursor mass consistency | Candidate sequence fits the precursor mass constraint | Different candidates can share the same total mass | Review rank separation |
| Few unexplained peaks | The candidate explains most dominant spectral evidence | Mixed spectra can hide another peptide | Recheck isolation purity |
| Long sequence tag | Core region is directly supported | Flanking residues may still be inferred | Target uncertain ends in follow-up |
The practical takeaway is straightforward: a strong report usually combines several of these features. One favorable metric by itself rarely settles the sequence.
Service Routes to Consider
For this project scenario, readers usually compare these service routes before requesting a quote or submitting samples.
Where Confidence Breaks Down
Four uncertainty sources show up in most real review decisions.
Incomplete fragmentation
A spectrum does not need to be perfect to be useful, but missing backbone cleavages force the algorithm to bridge unsupported intervals. Those intervals may still yield a reasonable candidate sequence, yet they should not be treated as equal in confidence to directly observed ladder segments.
PTM-related ambiguity
A post-translational modification changes the expected mass difference and can create competing interpretations. An unexpected gap may reflect a modified residue, a different residue combination, or a false assignment. When fragment ions do not localize the PTM cleanly, both sequence confidence and modification placement confidence may remain partial.
Isobaric residues
Leucine/isoleucine ambiguity remains a persistent limitation because these isobaric residues have the same mass in standard MS/MS-based workflows. Some near-isobaric substitutions can also stay difficult in poorly fragmented regions. A reported residue at such a site may therefore represent the best-supported class assignment rather than a uniquely proven amino acid identity.
Co-fragmentation and weak rank separation
If more than one precursor contributes to the fragmentation spectrum, the peak list contains mixed evidence. That raises the number of unexplained peaks and increases false assignment risk. Even without obvious contamination, a spectrum with weak rank separation between top candidates shows that the algorithm does not strongly favor one path over another.
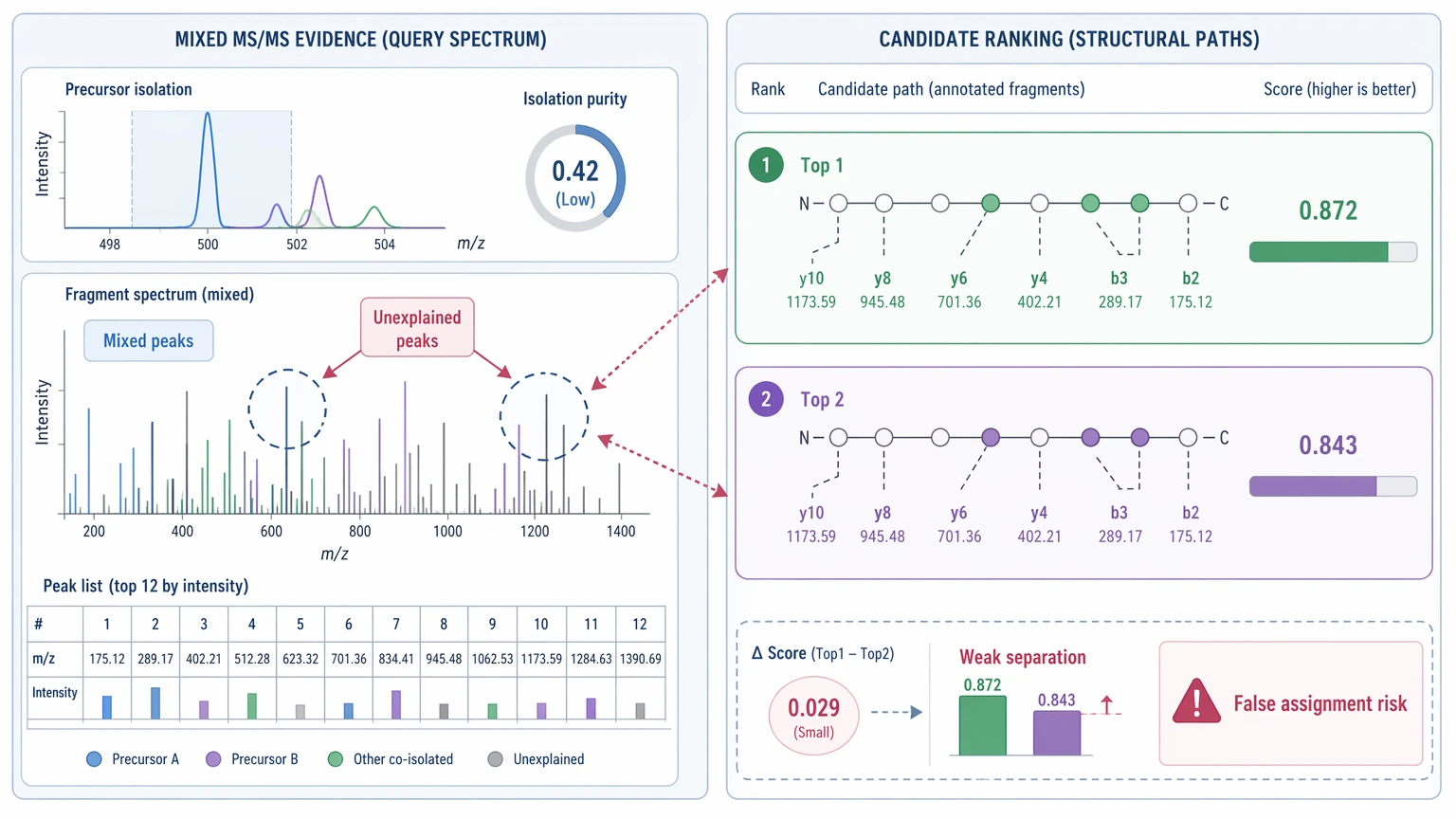
Expected Results and Validation Methods
The most useful de novo sequencing deliverables separate immediate output from follow-up confirmation instead of collapsing both into one confidence label.
Immediate deliverables usually include a ranked candidate sequence list, residue-level confidence across positions, sequence-level confidence, precursor mass agreement, annotated fragment ion assignments, and notes on unexplained peaks or ambiguous residues. For project review, these items show whether the sequence is close to actionable, better treated as a sequence tag, or too uncertain to interpret on its own.
Follow-up confirmation asks a different question: does the candidate hold up under an orthogonal test? Appropriate confirmation may include targeted LC-MS/MS on the same peptide, synthetic peptide matching, additional fragmentation acquisition, homology-based comparison, Edman-related confirmation in suitable contexts, or broader protein-level validation workflow design. In PTM-bearing peptides, the next step may need to separate backbone confirmation from PTM localization rather than force both questions into one pass.
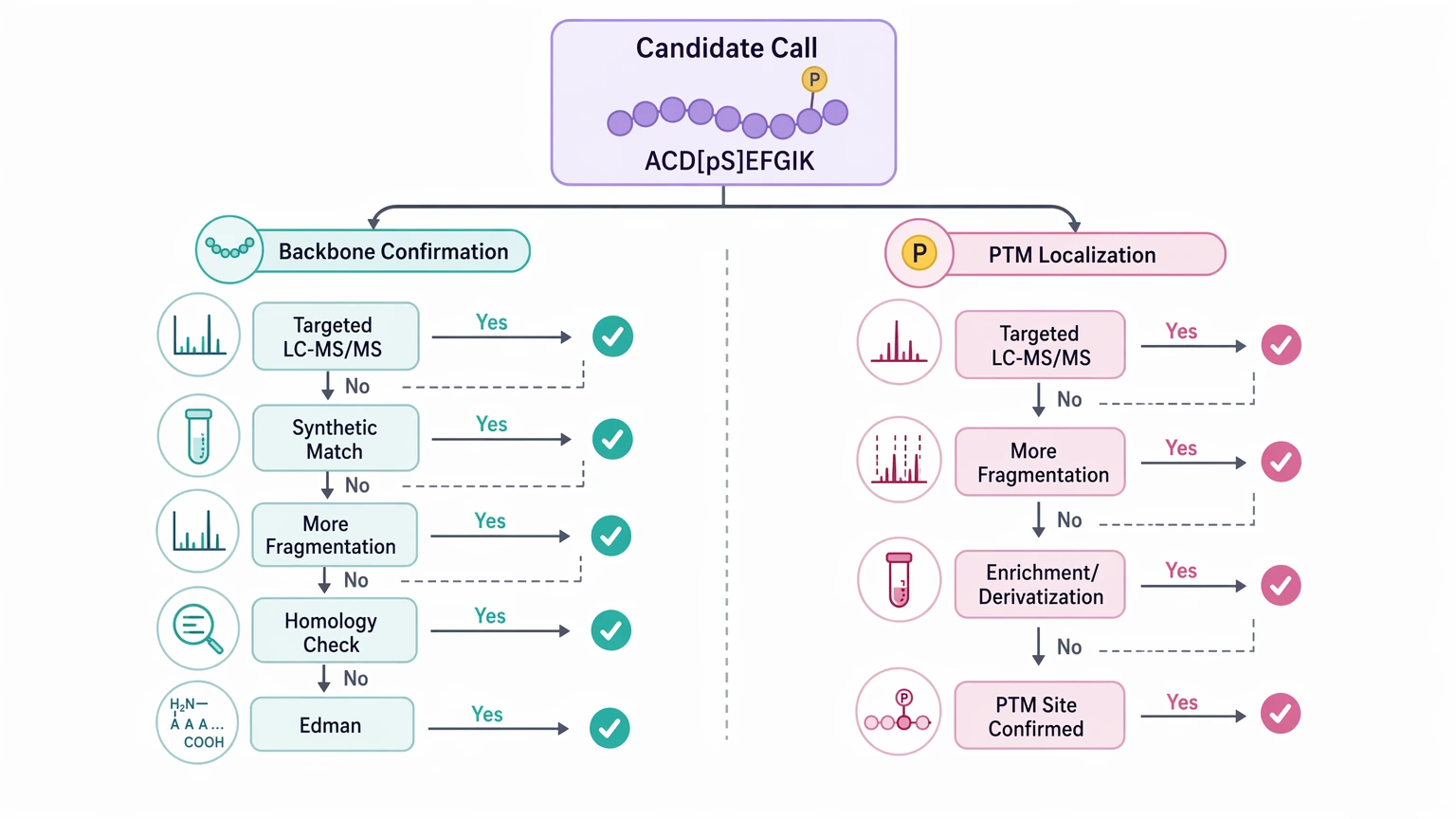
If your team needs to judge whether unknown-sequence spectra, PTM burden, and downstream confirmation goals fit a de novo workflow, submit your requirements to MtoZ Biolabs to evaluate your project and discuss an appropriate validation workflow before committing to a reporting standard.
Key Cautions and Practical Limits
Several limits deserve review before acting on a sequence call.
Reading the Output in Project Context
A useful de novo report does not promise universality. It helps researchers decide what can be said now and what still needs testing. In practice, the strongest output is often a peptide with solid internal sequence evidence, bounded ambiguity at a few positions, and a clear plan for targeted confirmation. That is enough for many discovery and characterization workflows, especially when database coverage is limited but downstream validation remains feasible.
For unknown peptides, PTM-burdened analytes, and reference-poor projects, the next step is usually a validation workflow matched to the exact uncertainty pattern in the report. If you are preparing that decision, contact us with the sample context, spectral constraints, and downstream objective so MtoZ Biolabs can help scope the project route before additional sequencing or confirmation work begins.
FAQ
Can de novo peptide sequencing still help when the full peptide cannot be reconstructed?
Yes. A strong sequence tag can still support homology searches, motif assignment, targeted assay design, or follow-up purification, even when one terminus or an internal region remains uncertain.
What should I look for first in a vendor or internal report?
Start with the annotated fragmentation spectrum, not the headline score. Check whether the proposed candidate sequence explains the dominant peaks, whether uncertain positions are marked clearly, and whether alternative candidates remain close in score.
Does a longer peptide automatically make de novo interpretation harder?
Often yes, because longer peptides are more likely to show incomplete fragmentation, internal gaps, and multiple plausible path extensions. That does not make them uninterpretable, but confidence usually becomes less uniform across the full backbone.
When is repeating LC-MS/MS more useful than moving directly to synthesis?
Repeat acquisition is usually the better first move when the original spectrum contains many unexplained peaks, weak ladder continuity, or signs of co-fragmentation. Synthesis becomes more efficient once the main uncertainty has been narrowed to a small number of positions.
Can de novo peptide sequencing answer a protein-level question by itself?
Only in a limited sense. Peptide-level de novo calls can support broader protein inference, but they do not automatically establish complete protein sequence, isoform structure, or proteoform architecture without additional peptide coverage and confirmation steps.
Are confidence scores comparable across different de novo software tools?
Not always. Different tools define scoring function components differently, so the more reliable comparison is usually inside the same workflow: top versus lower-ranked candidates, local confidence patterns, and agreement with the annotated spectral evidence.
How to order?

