





Co-IP-MS vs XL-MS: Which Protein Interaction Workflow Should You Choose?
-
The bait protein has a validated antibody or can be expressed with a tag.
-
The target complex is expected to remain stable during lysis and washing.
-
The project needs a ranked candidate list for follow-up validation.
-
The biological condition matters, such as treatment, mutation, time point, or cell type.
-
The study can include negative controls, input samples, and replicate experiments.
-
The complex or candidate interaction is already defined.
-
The project needs contact-site evidence rather than a broad partner list.
-
The protein complex can tolerate cross-linking without major disruption.
-
Structural models, cryo-EM maps, AlphaFold predictions, or domain maps need experimental restraints.
-
The sample amount and purity are sufficient for low-abundance cross-linked peptide detection.
Protein interaction studies often start with a practical problem: the biology points to a complex, pathway, receptor, or regulatory protein, but the experimental question is not yet clear enough to choose a workflow. One project may need to discover unknown binding partners from a native- like cellular context. Another project may already have a defined complex and need residue-level distance information to support a structural model. Co-IP-MS and XL-MS both use mass spectrometry, but the two workflows answer different questions. Choosing the wrong workflow can lead to weak enrichment, confusing background proteins, low cross-linked peptide recovery, or data that does not support the next validation step.
The most useful way to compare Co-IP-MS vs XL-MS is to start with the desired readout. Co-IP-MS is usually stronger for bait-centered partner discovery and complex profiling. XL-MS is usually stronger for mapping spatial proximity within or between proteins. Both workflows can be valuable, but each workflow has different sample requirements, controls, failure modes, and interpretation limits.
| Customer Need | Recommended Service Direction |
| Need to discover proteins that co-purify with a bait | Co-Immunoprecipitation Protein Interaction Analysis Service |
| Need residue-level proximity or contact- site evidence | Chemical Cross-Linking Mass Spectrometry Analysis Service |
| Need a broader MS-based protein interaction workflow | MS-Based Protein-Protein Interaction Analysis Service |
| Need bait enrichment without antibody- based Co-IP | Pull Down based Protein Analysis Service with Mass Spectrometry |
For teams comparing interaction workflows before sample submission, MtoZ Biolabs can help match Co-IP-MS, XL-MS, or pull-down MS to the biological question, sample type, and expected data output.
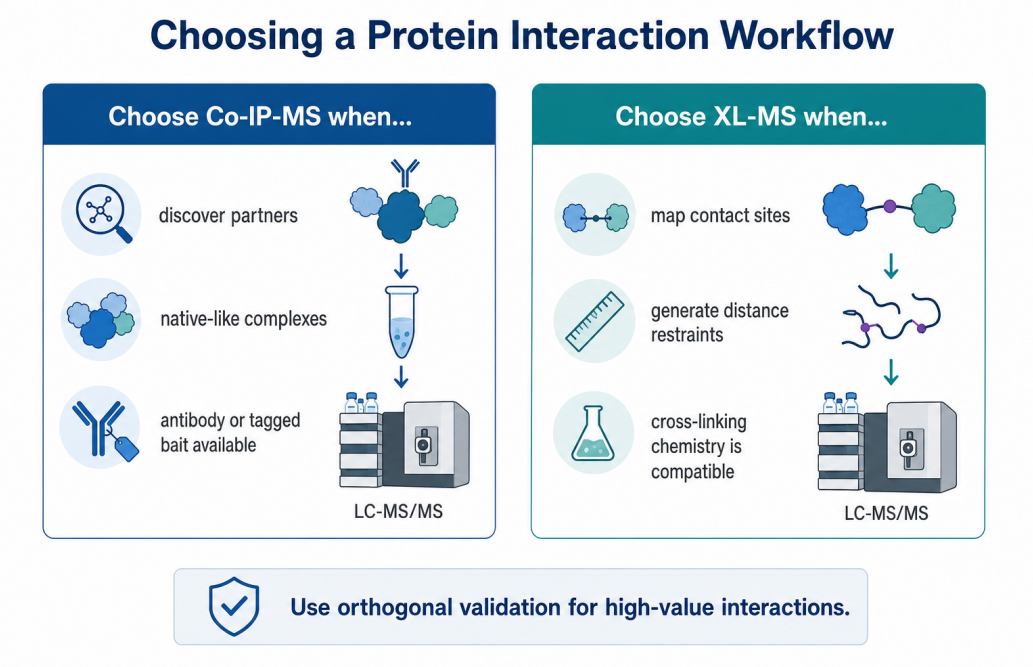
Figure 1. A practical decision map for choosing between Co-IP-MS and XL-MS.
What Co-IP-MS and XL-MS Actually Measure
Co-IP-MS, or co-immunoprecipitation followed by mass spectrometry, enriches a bait protein and associated proteins from a cell lysate or other biological matrix. The mass spectrometer identifies proteins that co-purify with the bait. A strong Co-IP-MS design can reveal direct interactors, complex members, pathway-associated proteins, and condition-dependent binding partners. However, Co-IP-MS does not prove direct physical contact by itself. A protein may appear because the protein binds indirectly through a larger complex, associates with beads, or appears as a recurring background contaminant.
XL-MS, or cross-linking mass spectrometry, uses a chemical cross-linker to covalently connect amino acid residues that are close in space. After digestion and LC-MS/MS analysis, cross-linked peptides provide distance restraints. These restraints can support protein interface mapping, conformational analysis, and structural modeling. XL-MS can be performed on purified complexes and, in selected cases, more complex samples. The method is powerful, but cross-linked peptides are usually low-abundance and technically challenging to identify with high confidence.
The core difference is simple: Co-IP-MS asks, "Which proteins are enriched with the bait?" XL-MS asks, "Which regions are close enough to be linked?" That distinction should guide the entire decision.
Key Comparison Dimensions
A fair comparison should not treat Co-IP-MS and XL-MS as interchangeable discovery tools. The workflows differ across at least four dimensions: biological question, sample state, data output, and validation burden.
| Dimension | Co-IP-MS | XL-MS |
| Primary question | Which proteins co-purify with the bait? | Which residues or regions are close in space? |
| Best application | Partner discovery, complex profiling, condition comparison | Interface mapping, topology analysis, structural restraints |
| Main dependency | Antibody specificity, bait expression, lysis conditions, controls | Cross-linker chemistry, complex stability, peptide coverage, database quality |
| Typical output | Protein list, enrichment score, spectral or intensity evidence | Cross-linked peptide pairs, distance constraints, residue-level proximity evidence |
| Main limitation | Indirect binders and background proteins require careful filtering | Lower peptide recovery and more complex data analysis |
Co-IP-MS is often the better first experiment when the interaction network is unknown. The workflow can screen a wider set of candidate partners, especially when a reliable antibody or tagged bait is available. XL-MS is more suitable when the protein complex is already known or strongly suspected, especially when the project needs structural information that a simple interactor list cannot provide.
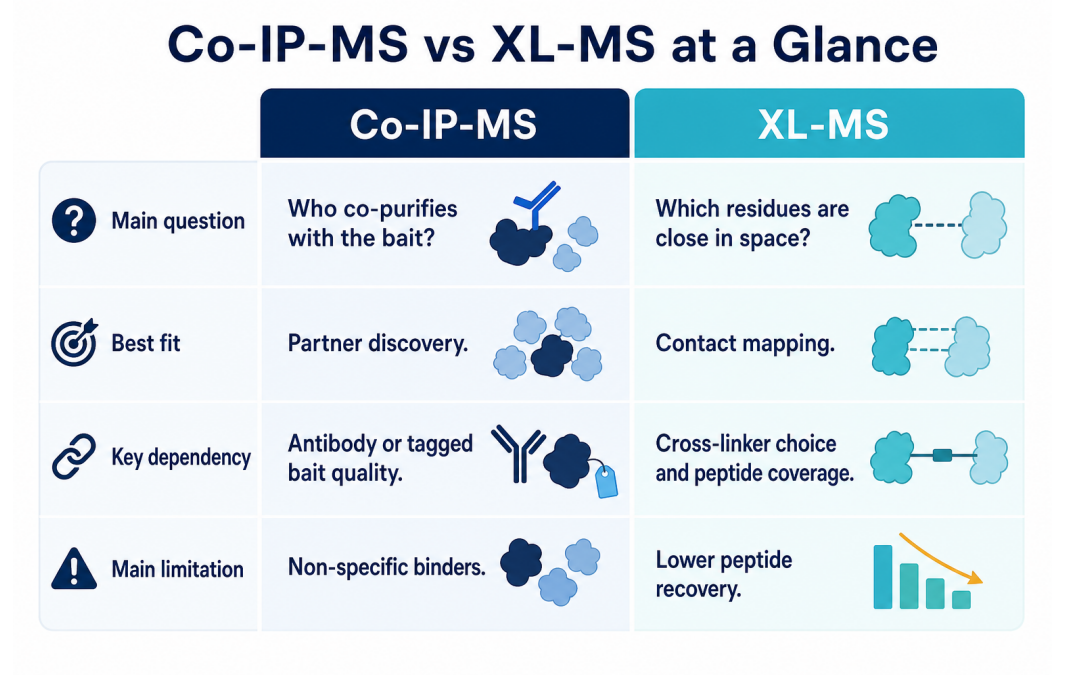
Figure 2. Co-IP-MS and XL-MS differ in question, best fit, dependency, and limitation.
When Co-IP-MS Is the Better Choice
Co-IP-MS is a strong option when the project goal is to discover or compare protein partners under biologically relevant conditions. For example, a researcher may want to compare the interactome of a transcription factor before and after stimulation. A drug discovery team may want to identify proteins that co-purify with a tagged target under treated and untreated conditions. In these cases, Co-IP-MS provides a practical entry point because the workflow can capture complex-level association rather than only direct residue contact.
Co-IP-MS is especially useful when:
The main risk is overinterpreting the protein list. Co-purification is not the same as direct binding. Co-IP-MS data should be interpreted with enrichment statistics, known contaminant databases, control pulldowns, pathway context, and orthogonal validation. Western blot, reciprocal Co-IP, targeted MS, functional assays, or imaging-based colocalization may be needed for high-value candidates.
When XL-MS Is the Better Choice
XL-MS is usually the better choice when the project needs spatial information. The method can reveal which residues or regions are close enough to be chemically linked. This makes XL-MS valuable for protein complex topology, interface analysis, conformational comparison, and model restraint generation.
XL-MS is especially useful when:
XL-MS does not usually replace Co-IP-MS for early discovery. Instead, XL-MS can deepen a project after discovery by adding structural context. For instance, Co-IP-MS may identify a candidate protein complex, while XL-MS may help determine which domains are close enough to support a mechanistic model.
Sample Requirements and Expected Outputs
Sample planning is one of the most important differences between the two workflows. Co-IP-MS can work from cells, tissues, or lysates if the bait enrichment is specific and the complex survives extraction. XL-MS often benefits from a cleaner, more defined sample because cross-linked peptide identification becomes harder as sample complexity increases.
| Planning Factor | Co-IP-MS Consideration | XL-MS Consideration |
| Sample state | Cell lysate, tissue lysate, or compatible biological matrix | Purified complex, enriched complex, or compatible lysate |
| Critical reagent | High-specificity antibody, tagged bait, or affinity resin | Suitable cross-linker with appropriate spacer length and reactive groups |
| Controls | IgG control, bead-only control, tag- only control, input, biological replicates | Non-cross-linked control, cross-linker titration, digestion control, replicate LC- MS/MS |
| Output format | Interactor list with enrichment and confidence ranking | Cross-linked peptide pairs with residue positions and distance information |
| Common QC concern | Non-specific binding and bait recovery | Cross-linking efficiency and cross-linked peptide identification rate |
For Co-IP-MS, sample lysis must preserve the target interaction while reducing non-specific binding. Harsh detergents may disrupt weak or transient complexes. Mild buffers may preserve interactions but increase background. For XL-MS, cross-linker selection and reaction conditions are central. Over-cross-linking may reduce peptide recovery, while under-cross-linking may produce too few informative links.
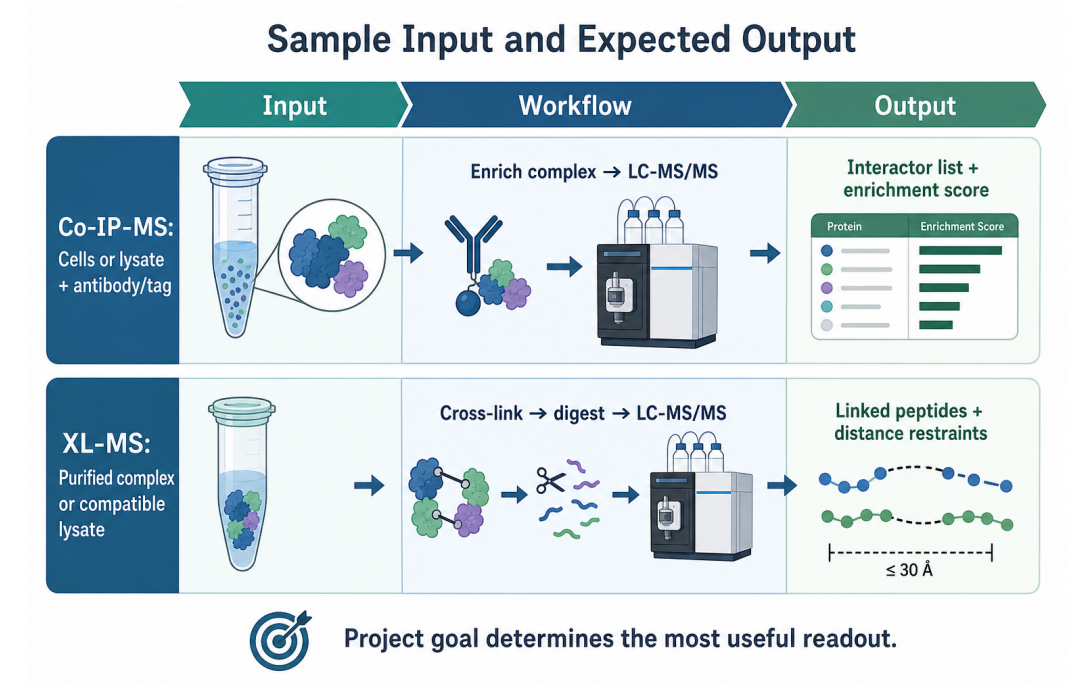
Figure 3. Sample input and expected output differ between Co-IP-MS and XL-MS.
Decision Recommendations by Research Goal
Choose Co-IP-MS when the main goal is discovery. If the project starts with a bait protein and a broad biological question, Co-IP-MS can provide a candidate interactor list. This is often the right first step for pathway mapping, disease mechanism exploration, mutation comparison, or treatment-response studies.
Choose XL-MS when the main goal is structural interpretation. If the project already has a defined complex, XL-MS can provide residue-level proximity evidence that supports domain mapping, interface analysis, or model refinement. This is particularly useful when computational predictions need experimental constraints.
Use both workflows when the project needs discovery and mechanism. A common strategy is to run Co-IP-MS first to identify candidate partners, then apply XL-MS to a prioritized complex. This staged design reduces risk because XL-MS is applied to a better-defined target. It also prevents the project from expecting one workflow to answer every question.
If the project has limited sample amount, uncertain antibody performance, or a weakly expressed bait, feasibility testing is important before committing to a full workflow. In many cases, a small pilot can reveal whether the enrichment or cross-linking chemistry is strong enough for confident LC-MS/MS analysis.
Common Pitfalls and How to Avoid Them
One common Co-IP-MS mistake is using insufficient controls. A single bait pulldown without an IgG, bead-only, tag-only, or condition-matched control can produce a long list of proteins that looks impressive but is hard to interpret. Controls are not optional because background proteins can dominate affinity enrichment experiments.
Another mistake is expecting XL-MS to generate a complete interaction map from a complex sample. XL-MS evidence is sparse by nature. The absence of a cross-link does not prove the absence of contact. Peptide detectability, digestion efficiency, residue availability, and instrument duty cycle all affect the observed result.
A third mistake is ignoring the next validation step. Co-IP-MS candidates may need reciprocal Co- IP, targeted proteomics, or functional validation. XL-MS links may need structural modeling, mutagenesis, or orthogonal biophysical evidence. The best workflow is the one that produces data aligned with the next decision.
FAQ
1. Is Co-IP-MS better than XL-MS for discovering new protein partners?
Usually yes. Co-IP-MS is generally better suited for discovering proteins that co-purify with a bait in native-like conditions. XL-MS can identify contacts, but it is usually more efficient when the candidate complex is already defined.
2. Can XL-MS prove a direct protein-protein interaction?
XL-MS provides strong proximity evidence when cross-linked residues are confidently identified. The evidence can support direct or near-direct contact, but interpretation still depends on cross- linker length, protein flexibility, sample purity, and data confidence.
3. Should Co-IP-MS and XL-MS be used together?
They can be highly complementary. Co-IP-MS can identify candidate partners, and XL-MS can add residue-level spatial information for prioritized complexes. This combined strategy is useful for mechanism-focused studies.
4. What is the biggest risk in Co-IP-MS data interpretation?
The biggest risk is treating all enriched proteins as direct binders. Co-IP-MS results require controls, enrichment analysis, contaminant filtering, biological context, and follow-up validation.
5. What is the biggest risk in XL-MS experiments?
The biggest risk is low cross-linked peptide recovery. Poor cross-linker selection, insufficient sample quality, incompatible buffers, and incomplete database preparation can reduce useful identifications.
Conclusion
Co-IP-MS and XL-MS are both valuable protein interaction workflows, but they serve different experimental goals. Co-IP-MS is usually the better choice for bait-centered partner discovery and condition-dependent complex profiling. XL-MS is usually the better choice for contact mapping, distance restraints, and structure-aware interpretation. The decision should be based on the biological question, sample state, reagent quality, expected output, and validation plan.
For interaction projects that need a clear workflow decision before sample preparation, MtoZ Biolabs can provide technical consultation for Co-IP-MS, XL-MS, and MS-based protein- protein interaction analysis, helping researchers align the method with the data needed for publication, validation, or downstream mechanism studies.
How to order?

