





Bottom-Up Proteomics in Single-Cell Proteomics: Challenges, Workflow, and Method Selection
- Single mammalian cells contain only a few hundred picograms of protein, so adsorption loss and incomplete recovery can erase low-abundance peptides.
- Microfluidic chips, nanoliter liquid handling, TMT multiplexing, and carrier-boost strategies help scale single-cell bottom-up workflows.
- DIA and ion mobility-enhanced acquisition reduce stochastic missingness compared with DDA in sparse single-cell data.
- Missing values, normalization, and batch effects need dedicated handling because single-cell proteomics data are sparse and zero-inflated.
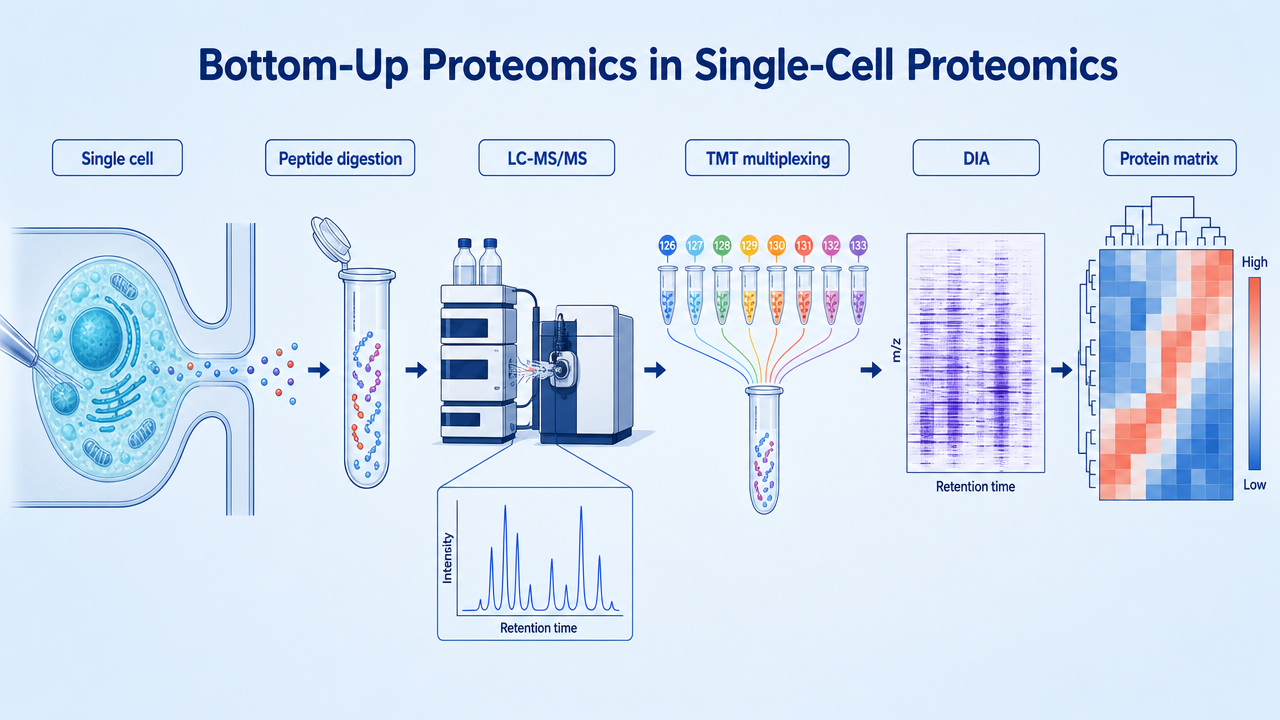
Bottom-up proteomics digests proteins into peptides in vitro, identifies them by LC-MS/MS, and infers parent proteins from peptide evidence. In single-cell proteomics, that workflow must work with picoliter sample volumes and picogram-level protein input, so sensitivity, sample loss control, separation efficiency, and quantitative consistency matter more than in bulk proteomics.
Key Takeaways
What Bottom-Up Single-Cell Proteomics Measures?
Bottom-up single-cell proteomics measures peptide and protein abundance in individual cells or very small cell populations. Unlike top-down proteomics, which analyzes intact proteins, the bottom-up route trades direct proteoform readout for higher throughput, mature software, and compatibility with labeling, carrier boost, and high-sensitivity LC-MS/MS platforms.
Related Services
Single-Cell Proteomics Analysis
Plant Single-Cell Proteomics Analysis
4D-DIA Quantitative Proteomics Service
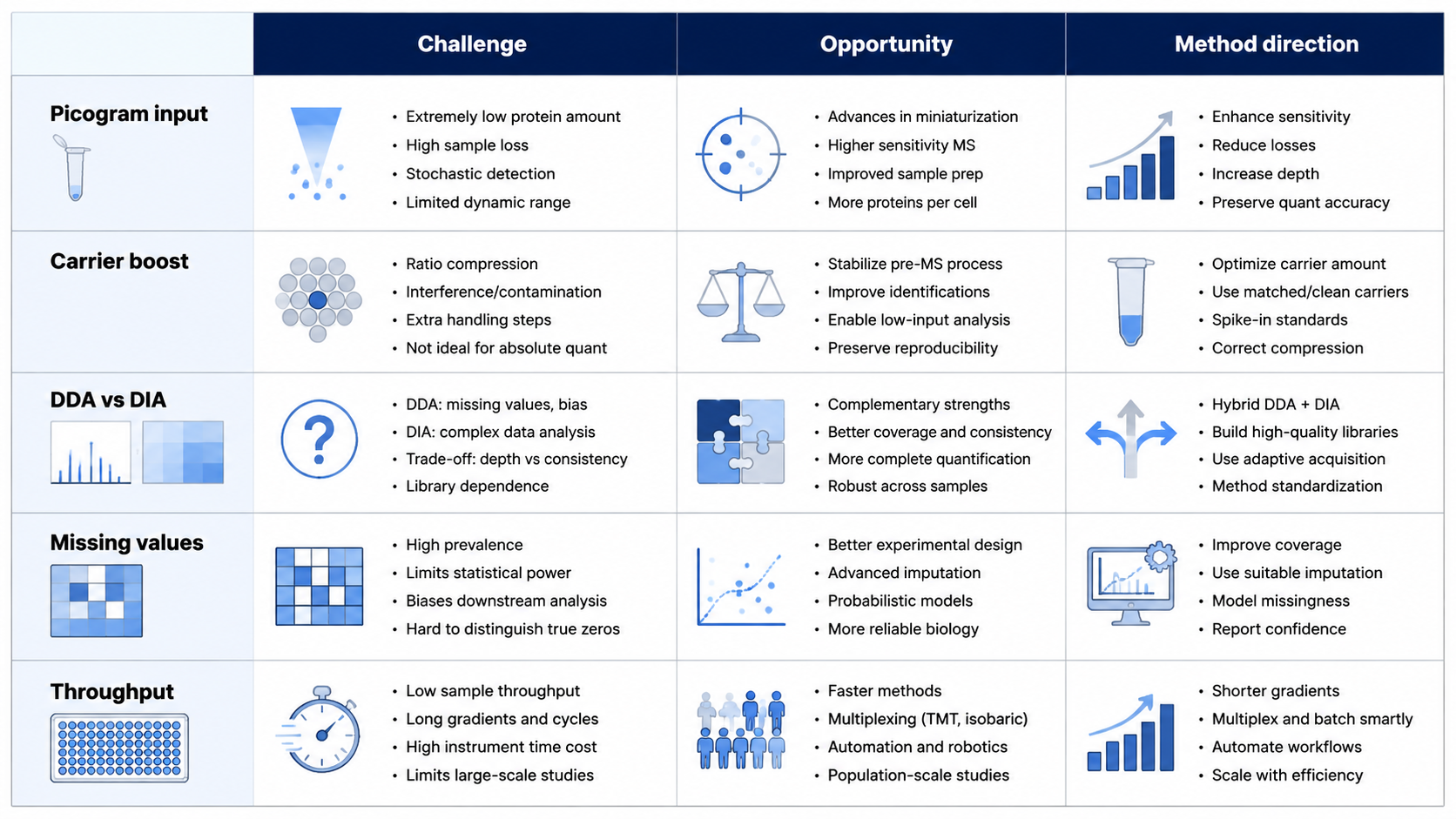
Sample Preparation Challenges
1. Efficient Lysis and Extraction
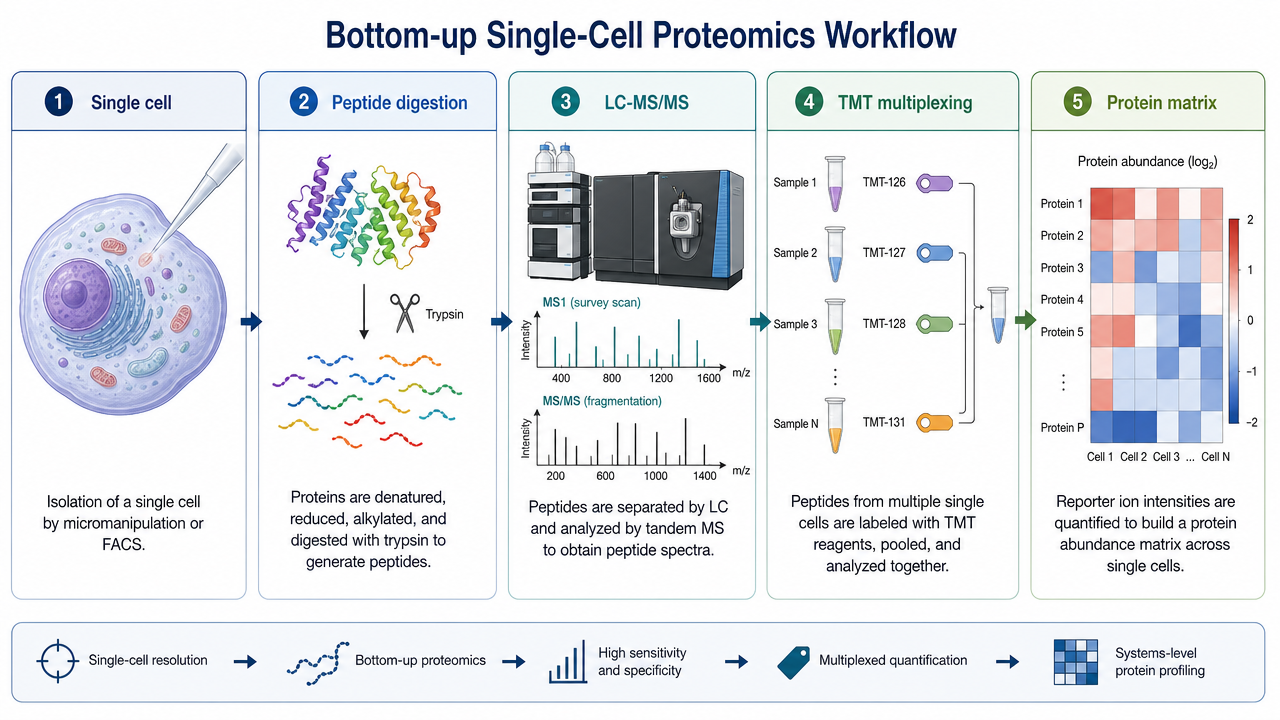
A single mammalian cell may contain only a few hundred picograms of protein. Any surface adsorption or handling loss can reduce peptide recovery enough to block detection. Microfluidic platforms that lyse and extract peptides in microwells, together with automated nanoliter-to-microliter liquid handling, reduce human error and improve reproducibility.
2. Multiplex Labeling and Carrier Boost
Single-cell proteomics often uses TMT isobaric labeling to multiplex many cells in one MS run. Carrier-boost strategies add more peptide material to improve MS1 signal and peptide-spectrum match rates. Too much carrier can mask cell-specific low-abundance peptides, so carrier-to-sample ratio and acquisition settings need tuning.
Separation and Detection
Nanoliter-scale LC is common in single-cell proteomics, but very low flow rates increase clogging and instability risk. Capillary electrophoresis-mass spectrometry offers zeptomole-level sensitivity. Ion mobility-enhanced DIA adds another separation dimension and can increase confidence in MS2 identifications.
DDA selectively fragments precursors and often misses low-abundance peptides. DIA samples fragment ions across the mass range and pairs well with machine learning spectral libraries for high-throughput single-cell studies.
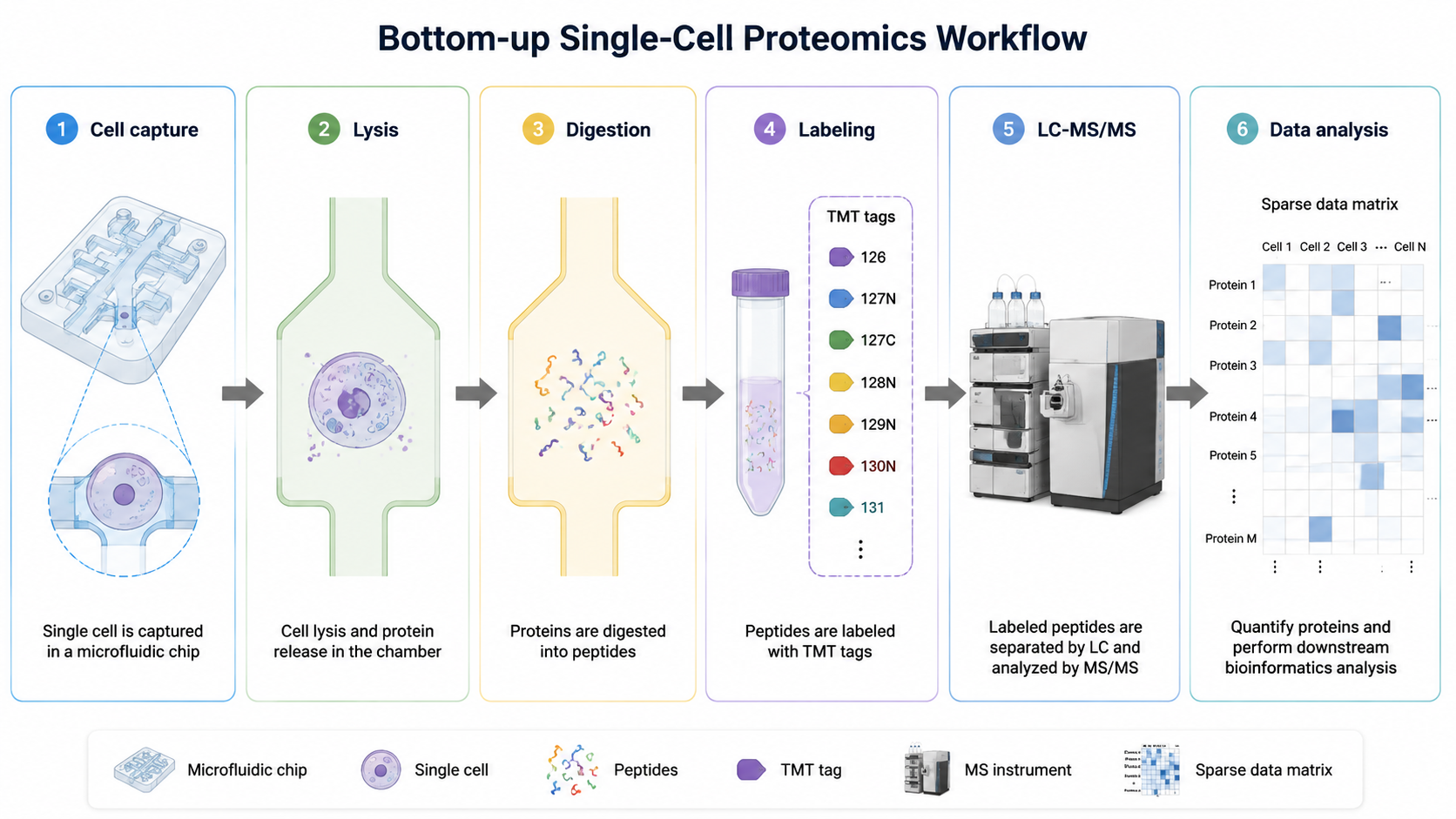
Data Analysis Challenges
Low-intensity MS/MS spectra increase search ambiguity. Stricter false discovery rate control and tools such as DIA-NN or Spectronaut can improve sensitivity. Missing values remain common, so carrier channels, imputation, and normalization must be chosen carefully. Experiments profiling thousands of cells need statistics suited to sparse, zero-inflated abundance tables.
Opportunities and Future Directions
| Challenge | Why it matters | Practical direction |
|---|---|---|
| Picogram input | Easy sample loss | Microfluidics and low-volume automation |
| Dynamic range | Low-abundance proteins disappear | Carrier boost with tuned ratios |
| Throughput vs depth | Long gradients reduce sample rate | Whisper-flow LC plus rapid DIA |
| Missing values | Weak statistics | DIA, carrier channels, careful imputation |
FAQ
1. What is bottom-up proteomics in single-cell proteomics?
It is the analysis of proteins in individual cells by digesting proteins into peptides, identifying them with LC-MS/MS, and reconstructing protein-level abundance from peptide evidence.
2. Why is single-cell bottom-up proteomics difficult?
Cells contain very little protein, peptide signals are sparse, sample handling losses are magnified, and quantitative data often contain many missing values.
3. What is carrier boost in single-cell proteomics?
Carrier boost adds extra peptide material to improve identification sensitivity. It can increase peptide-spectrum match rates but may dilute cell-specific low-abundance signals if overloaded.
4. Is DDA or DIA better for single-cell proteomics?
DIA is often preferred for high-throughput single-cell studies because it reduces stochastic missingness and supports more complete sampling across the mass range.
Conclusion
Bottom-up single-cell proteomics sits at the intersection of extreme sample scarcity and rapidly improving MS technology. Microfluidics, multiplex labeling, DIA, ion mobility, and better software are making single-cell protein profiling more practical for biological discovery.
How to order?

