





Bottom-Up Proteomics in Single-Cell Proteomics: Challenges, Solutions, and Opportunities
-
Single mammalian cells contain only a few hundred picograms of total protein, so surface adsorption and transfer loss can dominate the experiment.
-
Bottom-up workflows improve throughput, but they provide peptide-level evidence rather than intact proteoform information.
-
Microfluidics, nanoliter handling, TMT carrier channels, and low-flow separations reduce sample loss and improve identification depth.
-
DIA, ion mobility, and high-speed MS instruments reduce stochastic sampling and missing values in low-input datasets.
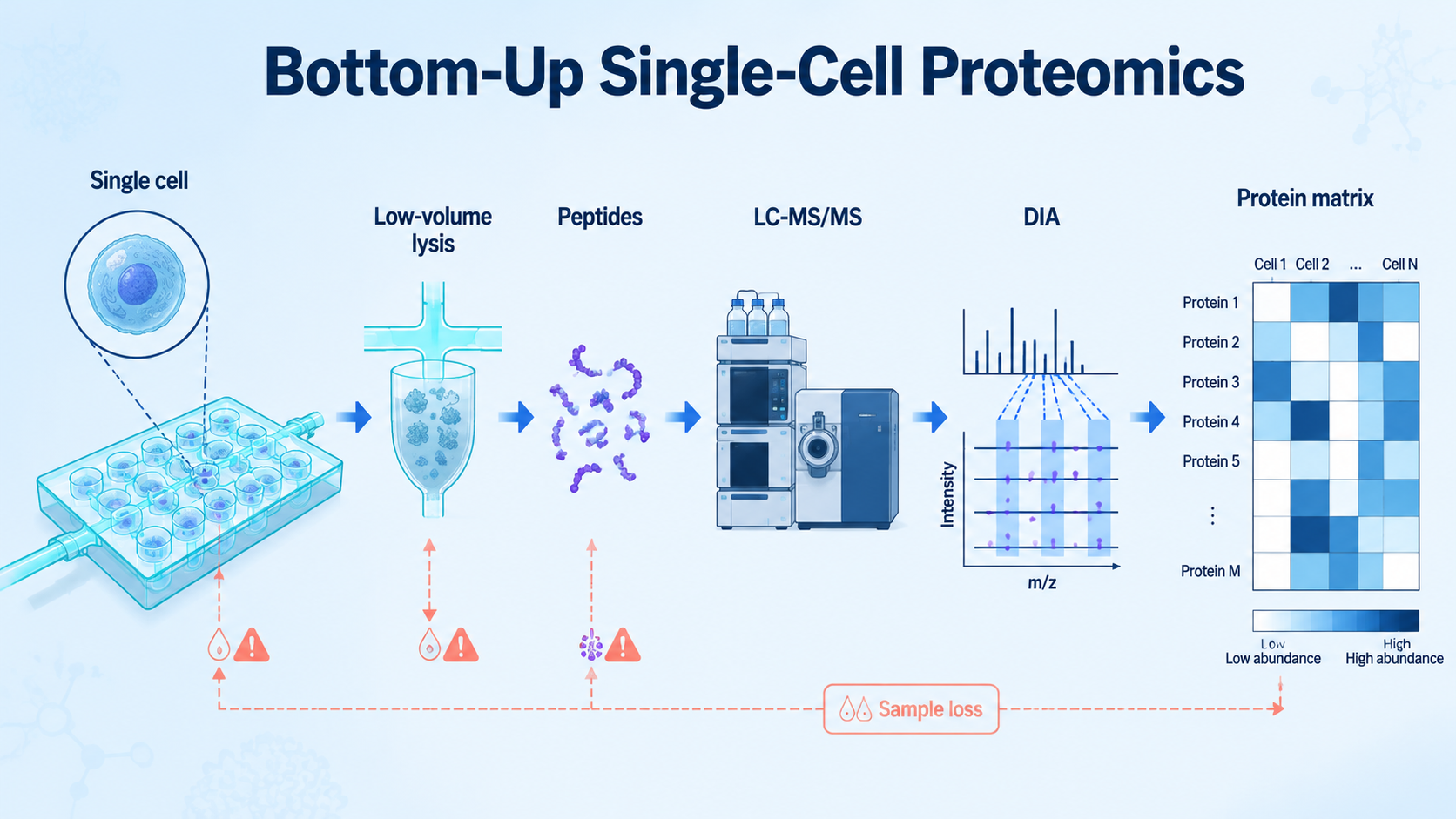
Bottom-up proteomics in single-cell proteomics digests proteins from individual cells into peptides, analyzes those peptides by LC-MS/MS, and reconstructs protein-level information from peptide evidence. The strategy is scalable and compatible with modern proteomics software, but it must work with picogram-level protein input, which makes sample loss, missing values, sensitivity, and quantitative bias the main technical barriers.
Key Takeaways
What Does Bottom-Up Single-Cell Proteomics Measure?
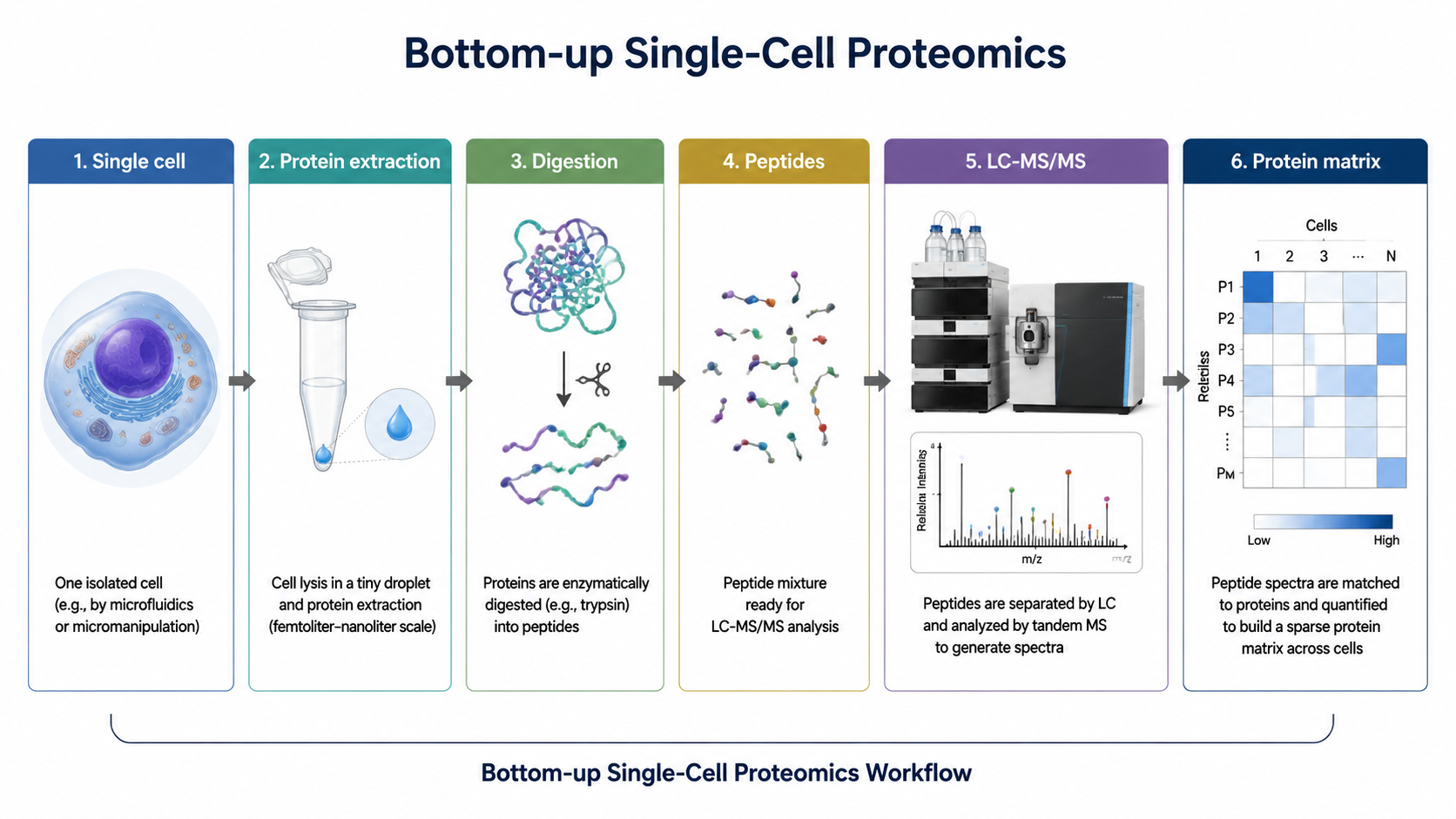
Bottom-up single-cell proteomics measures peptide signals derived from proteins in individual cells or very small cell populations. After digestion, peptides are separated, fragmented, identified, quantified, and mapped back to protein groups. The result is a protein abundance matrix that can reveal cell states, lineage differences, treatment responses, and pathway-level heterogeneity.
Related Services
Single-Cell Proteomics Analysis
Plant Single-Cell Proteomics Analysis
4D-DIA Quantitative Proteomics Service
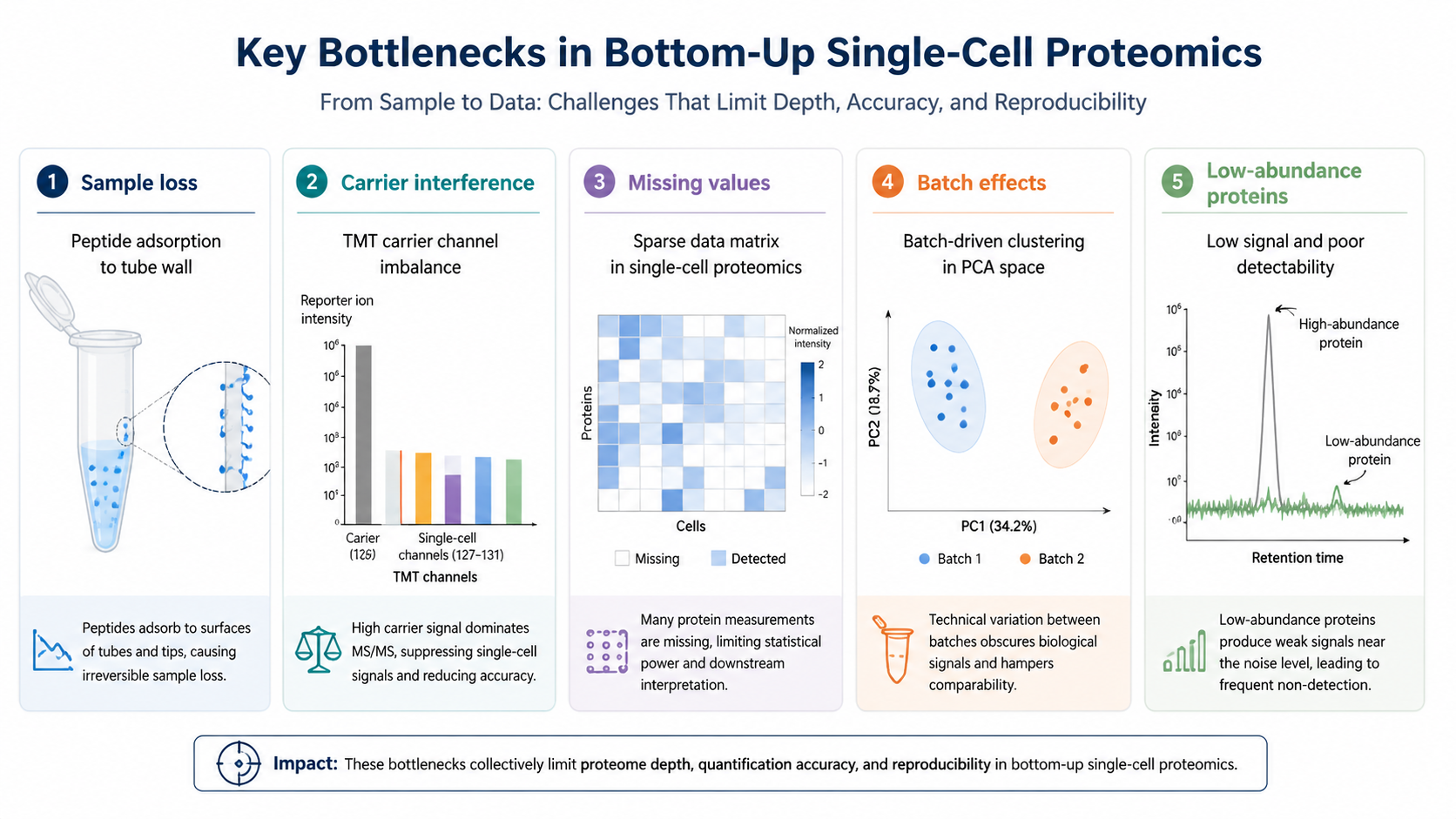
Challenge 1: Sample Loss at Picogram Input
At single-cell input, every surface matters. Protein and peptide adsorption to tubes, pipette tips, plates, and column materials can remove enough sample to change identification depth. Low-volume lysis, one-pot digestion, microfluidic chambers, and automated nanoliter handling are used to reduce transfer steps and exposed surfaces.
Sample preparation also has to balance cell lysis, digestion efficiency, and compatibility with LC-MS/MS. Detergents and salts may improve extraction but can suppress ionization or contaminate columns if they are not removed.
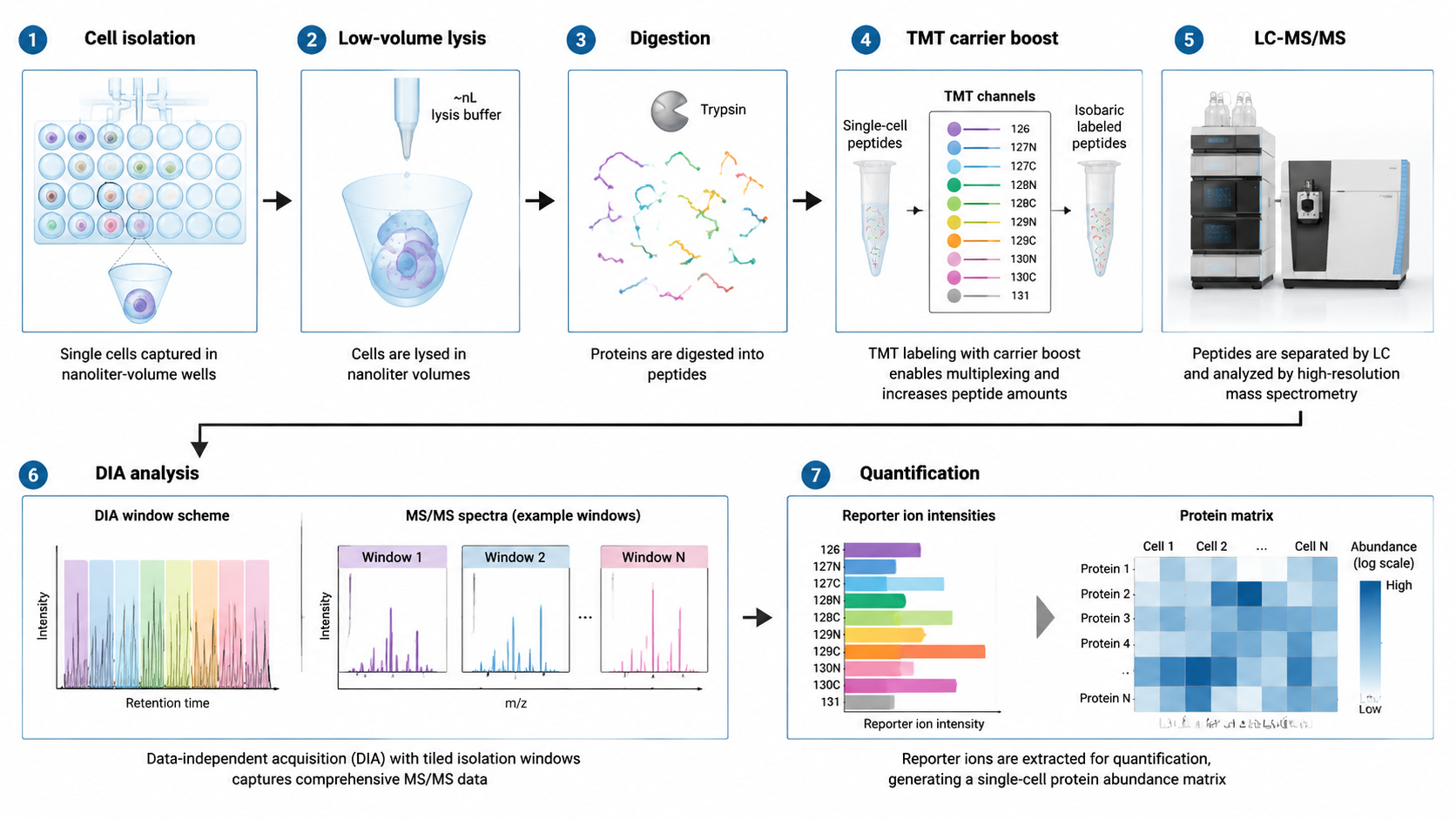
Challenge 2: Multiplexing and Carrier Boost
TMT-based multiplexing is widely used in single-cell proteomics because it allows multiple single cells to be combined into one MS run. Carrier channels add peptide material to improve MS1 signal and peptide identification. The trade-off is that too much carrier material can mask single-cell-specific low-abundance peptides or distort reporter ion quantification.
Challenge 3: Separation Sensitivity and Throughput
Nanoliter LC can improve sensitivity, but very low flow rates increase the risk of clogging, carryover, and run-to-run instability. CE-MS offers an orthogonal separation mechanism and can be useful for very low amounts of material or specific peptide classes. Ion mobility adds another separation dimension and can improve precursor resolution before fragmentation.
Challenge 4: DDA, DIA, and Missing Values
DDA can identify peptides deeply in some runs, but precursor selection is stochastic. In single-cell samples, where peptide signals are weak, stochastic selection can create many missing values. DIA samples broader m/z windows more systematically, which can reduce missingness and improve quantification across cells.
Challenge 5: Data Analysis and Statistics
Single-cell proteomics datasets are sparse and variable. Missing values may reflect low abundance, ion suppression, stochastic sampling, or technical loss. Normalization must handle differences in sample loading, batch, labeling channel, acquisition day, and cell state.
Opportunities
The strongest opportunities come from combining miniaturized sample preparation, robust LC or CE separation, DIA or ion mobility-enhanced acquisition, and machine learning-assisted data analysis. These improvements can make single-cell proteomics useful for cell-state mapping, tumor heterogeneity, rare cell populations, developmental biology, drug response, and clinical translation.
Method Selection
| Need | Useful Strategy | Why Does It Help | Main Trade-Off |
|---|---|---|---|
| Minimize sample loss | One-pot or microfluidic preparation | Reduces exposed surfaces and transfers | Requires workflow optimization |
| Increase peptide IDs | TMT carrier boost | Improves MS signal and PSM rate | Carrier can reduce single-cell specificity |
| Reduce missing values | DIA acquisition | More systematic peptide sampling | Needs strong analysis workflow |
| Improve separation | Low-flow LC, CE-MS, or ion mobility | Increases sensitivity and resolution | Can reduce throughput or increase complexity |
FAQ
1. What is bottom-up single-cell proteomics?
Bottom-up single-cell proteomics digests proteins from individual cells into peptides and analyzes them by LC-MS/MS to infer protein abundance and cell-state information.
2. Why is single-cell proteomics difficult?
Single cells contain picogram-level protein amounts. Sample loss, weak ion signals, missing values, and batch effects can strongly affect detection and quantification.
3. What is carrier boost in single-cell proteomics?
Carrier boost uses extra peptide material, often in a TMT carrier channel, to increase peptide identification rates. It must be balanced because excessive carrier can obscure single-cell-specific signals.
4. Is DIA useful for single-cell proteomics?
Yes. DIA can reduce stochastic precursor selection and improve cross-cell quantification, especially when paired with suitable software, predicted spectra, or ion mobility information.
Conclusion
Bottom-up proteomics gives single-cell proteomics a scalable analytical foundation, but the workflow has little tolerance for loss or bias. The best results come from treating preparation, acquisition, and data analysis as one system: protect the peptides, collect consistent spectra, and interpret sparse data with methods designed for low-input biology.
How to order?

