





4D Label-Free Quantitative Proteomics Workflow: From Sample Prep to Data Interpretation
- 4D label-free quantitative proteomics combines LC separation, m/z measurement, ion mobility, and intensity-based quantification.
- The workflow is only as strong as its sample preparation. Extraction, digestion, desalting, and batch design affect the final protein table.
- PASEF acquisition improves MS/MS sampling speed by coordinating trapped ion mobility separation with fragmentation.
- Label-free analysis avoids chemical labeling costs, but it requires careful normalization, QC tracking, and batch-effect control.
- Differential protein calls should be interpreted with peptide evidence, missing-value behavior, fold change, statistical confidence, and biological plausibility.

4D label-free quantitative proteomics measures protein abundance without isotope or isobaric labeling. The "4D" format adds ion mobility separation to conventional LC-MS/MS dimensions, so peptides are resolved by retention time, mass-to-charge ratio, ion mobility, and signal intensity. For researchers, the practical advantage is deeper peptide coverage and more reproducible quantification when the workflow is controlled from sample extraction through statistical analysis.
Key Takeaways
What is 4D Label-Free Quantitative Proteomics?
4D label-free quantitative proteomics is an LC-MS/MS workflow for comparing protein abundance across biological samples. It does not rely on SILAC, TMT, iTRAQ, or other labeling chemistry. Instead, relative abundance is inferred from peptide ion signals across runs.
In a 4D platform, each peptide is described by four information layers: retention time from liquid chromatography, mass-to-charge ratio from mass spectrometry, ion mobility or collisional cross section related information, and signal intensity used for quantification.
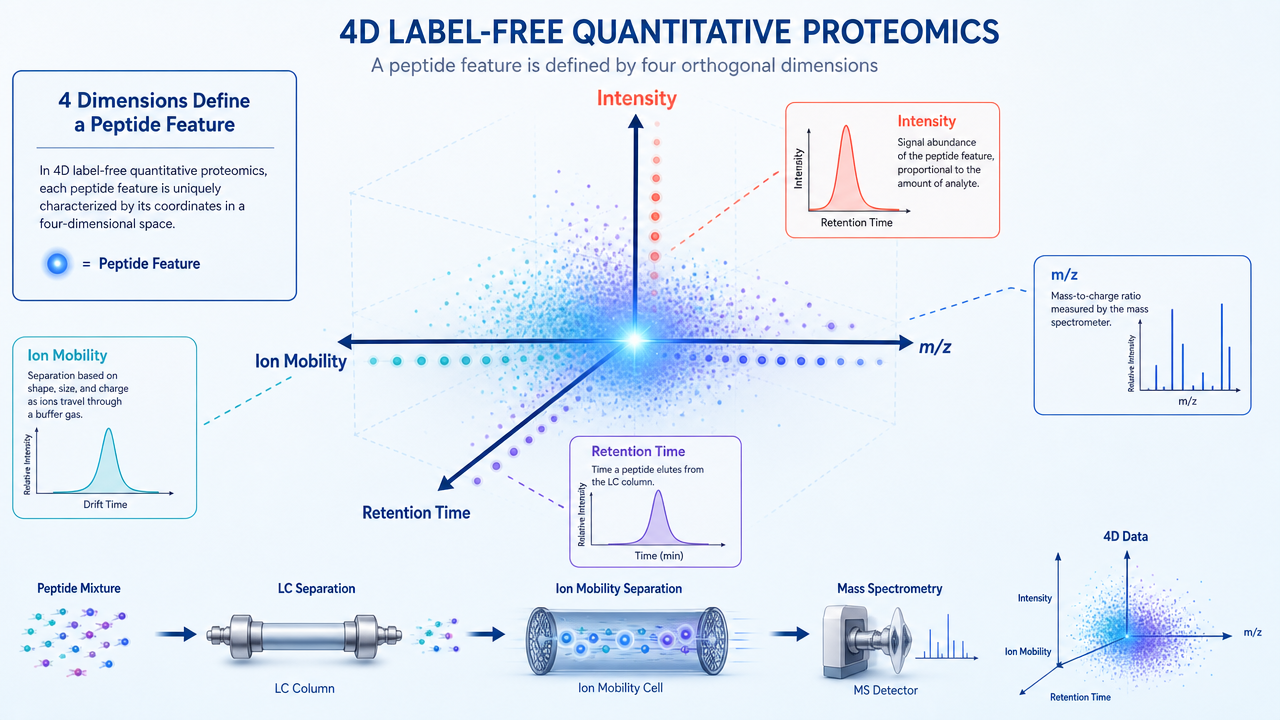
The added ion mobility dimension helps separate peptide features that overlap in conventional LC-MS/MS. That does not make the method automatic or immune to noise. It gives the data acquisition system more structure to work with.
Related Services
4D Label-free Quantitative Proteomics Analysis Service
4D Label-Free Quantitative Proteomics Service
Label-Free DIA Quantitative Proteomics
4D-DIA Quantitative Proteomics Service
Step 1: Experimental Design and Sample Planning
The workflow starts before protein extraction. Groups, replicates, randomization, sample amount, and QC placement should be defined before the first tube is processed. A technically clean LC-MS run cannot rescue a study where biological groups are underpowered or processed in a confounded batch order.
For discovery proteomics, biological replicates are more valuable than technical replicates. Technical repeats can diagnose instrument variability, but they do not capture biological variation. If tissue, plasma, serum, FFPE, exosome, or cell samples are used, the protocol should also define sample collection, storage, freeze-thaw history, and exclusion criteria.
Step 2: Protein Extraction and Quantification
Protein extraction should match the sample matrix. Cells and fresh tissue often use detergent or chaotrope-based lysis. Plasma and serum may need depletion, fractionation, or dynamic range management. FFPE samples require reversal of crosslinks and careful recovery of peptides or proteins.
After extraction, total protein is usually measured by BCA or another compatible assay. The goal is not just to know concentration. It is to load comparable material into digestion and to identify outlier samples before they consume MS time.
Step 3: Digestion, Cleanup, and Peptide Preparation
Proteins are typically reduced, alkylated, and digested with trypsin or a trypsin/Lys-C strategy. Digestion format may use in-solution digestion, FASP, S-Trap, SP3, or other cleanup-compatible approaches. The best choice depends on detergent content, sample amount, contaminants, and expected throughput.
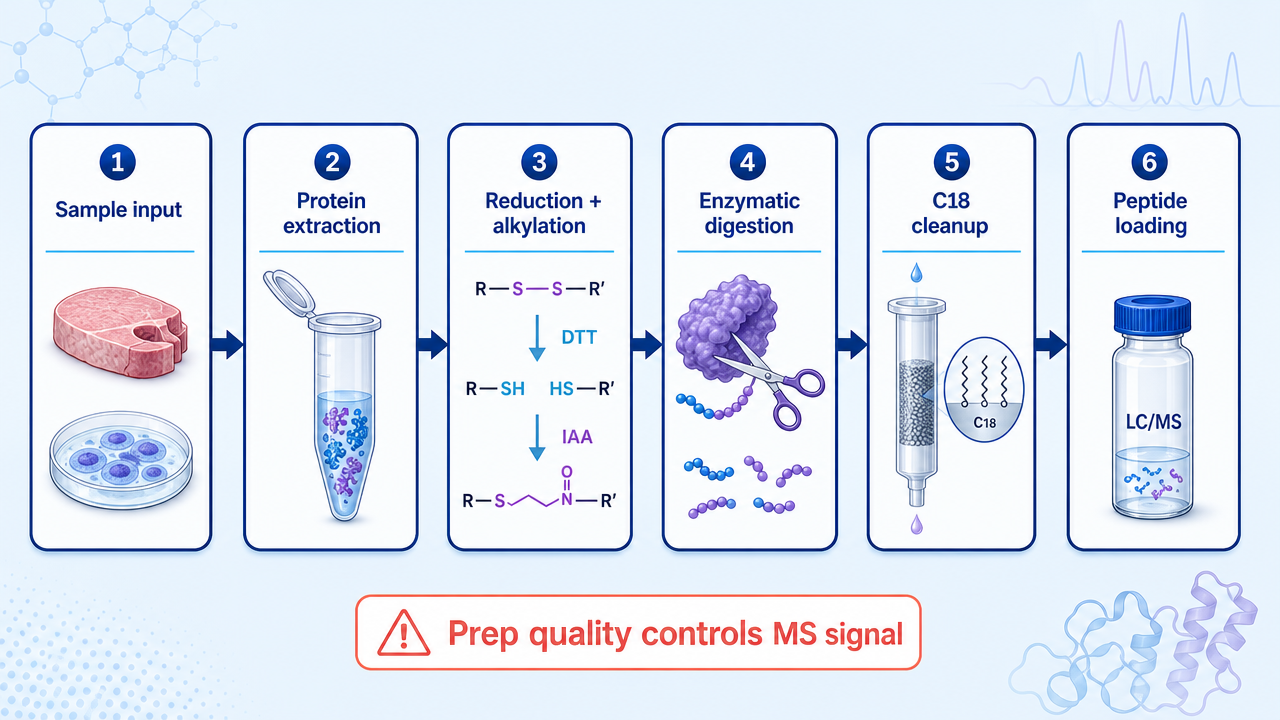
Desalting with C18 cleanup removes salts, detergents, and other contaminants that suppress ionization. Peptide amount, drying conditions, reconstitution solvent, and injection amount should be kept consistent across samples.
Step 4: LC-MS/MS Acquisition with Ion Mobility and PASEF
The LC system separates peptides by hydrophobicity before they enter the mass spectrometer. Nanoflow gradients are commonly used for deeper proteome coverage. Longer gradients can improve separation, but they reduce throughput. Shorter gradients increase throughput, but they put more pressure on acquisition speed and chromatographic reproducibility.
In 4D proteomics, ion mobility adds another separation step in the gas phase. On timsTOF platforms, PASEF coordinates ion accumulation, mobility separation, and fragmentation. The result is faster MS/MS sampling and better use of the ion beam compared with conventional sequential acquisition.
This is where 4D label-free analysis earns its name. The method does not simply collect more spectra. It organizes peptide evidence across retention time, m/z, ion mobility, and intensity, which improves feature detection and matching across runs.
Step 5: Database Search and Protein Inference
Raw data are processed with software such as MaxQuant, Spectronaut, DIA-NN, PEAKS, or platform-specific pipelines depending on acquisition mode and project design. The database search links MS/MS spectra or DIA signals to peptide sequences, then maps peptides to proteins.
Protein inference is a known source of ambiguity. Shared peptides can map to multiple isoforms or homologous proteins. For this reason, reports should distinguish protein groups, unique peptides, razor peptides, sequence coverage, and confidence thresholds.
Step 6: Label-Free Quantification and Normalization
Label-free quantification uses peptide ion intensity, extracted ion chromatograms, spectral evidence, or DIA feature signals to estimate relative abundance. Normalization then adjusts for systematic differences between runs, such as loading variation, total signal drift, or instrument response changes.
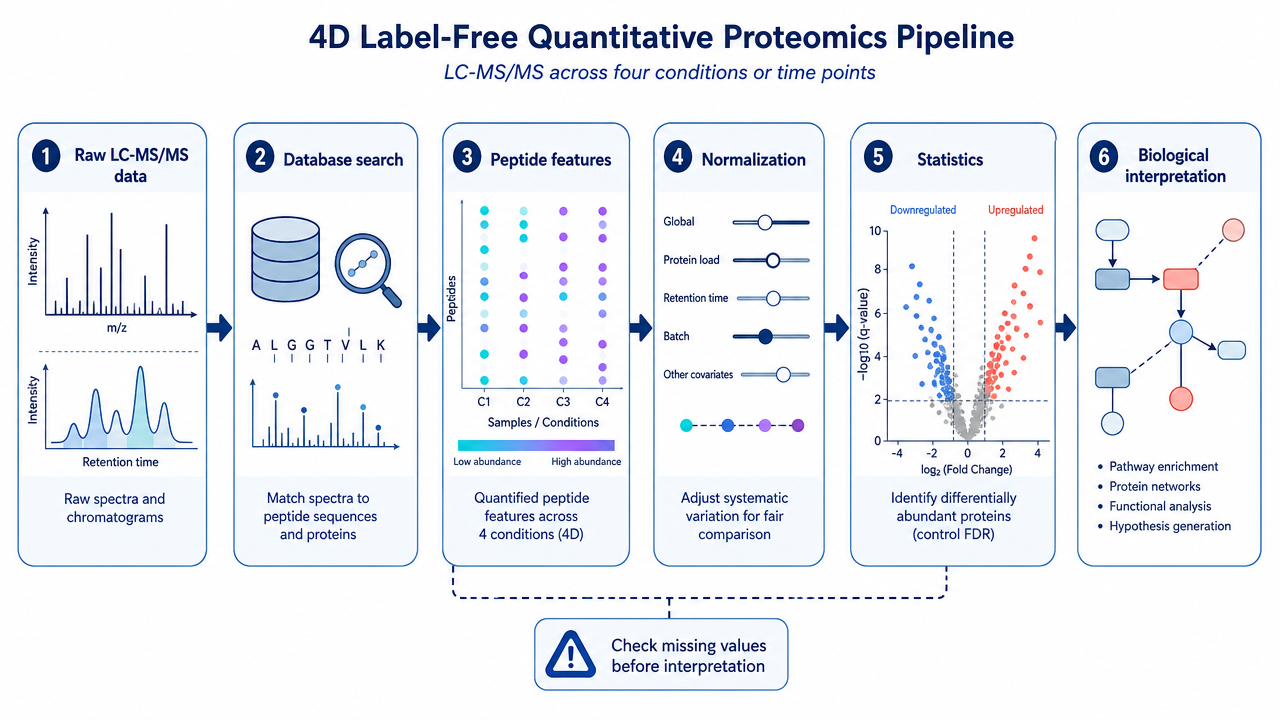
Missing values should be treated carefully. A missing value may reflect a low-abundance peptide, stochastic sampling, interference, matrix effects, or true absence. Imputing all missing values the same way can create artificial differences.
Step 7: Statistical Analysis and Biological Interpretation
Differential protein analysis usually combines fold change, p-value, multiple-testing adjustment, peptide evidence, and replicate consistency. A fold change threshold such as 1.5 or 2.0 can be useful, but it should not be used alone.
Common downstream analyses include volcano plots, heatmaps, principal component analysis, GO enrichment, KEGG pathway analysis, protein interaction networks, and clustering. These tools help interpret the protein table, but they do not replace validation. Candidate biomarkers or mechanism-related proteins often need PRM, Western blot, ELISA, immunostaining, or functional assays.
Main Advantages of 4D Label-Free Quantitative Proteomics
4D label-free proteomics is attractive because it avoids labeling chemistry while still supporting broad proteome discovery. It can compare many biological samples without designing labeling channels or mixing ratios in advance.
The ion mobility dimension helps resolve overlapping peptide features, and PASEF acquisition can improve MS/MS sampling efficiency. For limited samples, the method can provide useful proteome depth from relatively small peptide amounts, provided sample preparation is clean and losses are controlled.
The workflow is also flexible. It can be applied to cells, tissues, plasma, serum, cerebrospinal fluid, exosomes, FFPE material, and other matrices when extraction and cleanup are optimized for the sample type.
Main Disadvantages
Label-free workflows are sensitive to run-to-run variability. Instrument drift, column aging, sample loading differences, and batch effects can all influence intensity measurements. A robust QC design is therefore not optional.
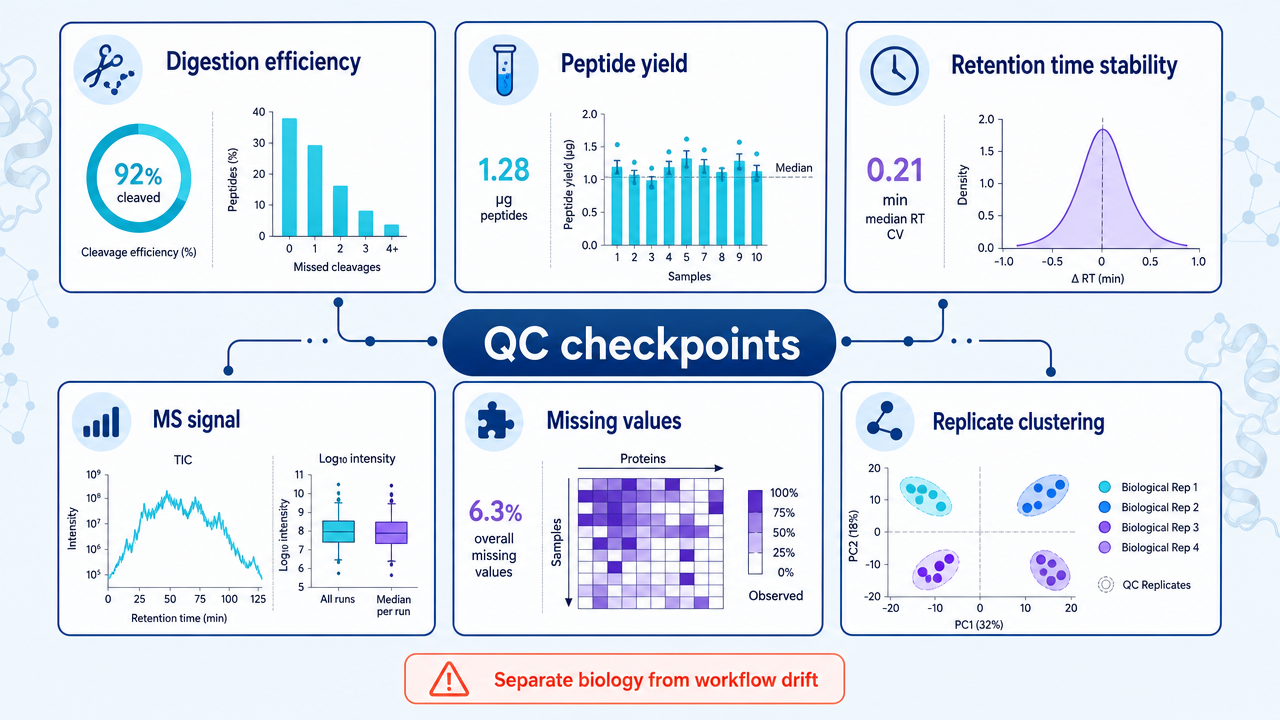
Dynamic range is another limitation. Plasma and serum contain highly abundant proteins that can mask lower-abundance signals. Tissue heterogeneity can also dilute cell-type-specific changes. The right workflow may require fractionation, depletion, enrichment, or a targeted follow-up assay.
Finally, label-free quantification gives relative abundance, not absolute concentration unless additional standards or calibration strategies are used.
4D Label-Free Proteomics versus Labeled Quantification
| Method | Best fit | Strength | Main tradeoff |
|---|---|---|---|
| 4D label-free LFQ | Broad discovery across many samples | No labeling chemistry, flexible design | More sensitive to run-to-run variation |
| DIA label-free proteomics | Reproducible large-cohort quantification | Strong data completeness and consistency | Requires optimized library or library-free workflow |
| TMT or iTRAQ | Multiplexed comparison in controlled batches | Samples are combined and measured together | Ratio compression and labeling cost |
| SILAC | Cell culture studies with metabolic labeling | Accurate relative quantification | Limited to compatible biological systems |
| PRM targeted proteomics | Validation of selected proteins or peptides | High specificity for candidates | Not a discovery-wide method |
FAQ
1. What does 4D mean in label-free quantitative proteomics?
In this context, 4D refers to retention time, mass-to-charge ratio, ion mobility, and signal intensity. These dimensions help separate and quantify peptide signals during LC-MS/MS analysis.
2. Is 4D label-free proteomics the same as DIA?
Not necessarily. "4D" describes the addition of ion mobility information, while DIA describes a data acquisition strategy. A project can use 4D-DIA, 4D-DDA, or other platform-specific workflows depending on the instrument and analysis plan.
3. How much sample is needed for 4D label-free proteomics?
Sample requirements depend on matrix, protein complexity, cleanup losses, and depth targets. Limited samples can be analyzed, but low input increases the need for careful extraction, digestion, desalting, and QC.
4. What are the main QC metrics for 4D label-free proteomics?
Useful QC metrics include peptide yield, digestion efficiency, identification number, MS1 signal stability, retention time reproducibility, ion mobility consistency, missing-value rate, replicate clustering, and QC sample drift.
5. When should label-free results be validated?
Validation is recommended when proteins support a biomarker claim, disease mechanism, drug target, pathway conclusion, or publication-critical result. PRM, Western blot, ELISA, and functional assays are common follow-up options.
Conclusion
4D label-free quantitative proteomics is a strong discovery workflow when the project needs broad protein coverage without labeling chemistry. Its performance depends on disciplined sample handling, clean peptide preparation, stable LC-MS/MS acquisition, and careful statistical interpretation. Used well, it turns complex biological samples into a protein-level map that can guide mechanism studies, biomarker discovery, and targeted validation.
How to order?

