





Why Peptide De Novo Sequencing Fails on Poor MS/MS Spectra and How to Improve Sequence Confidence
- you can trace a meaningful sequence tag from consecutive terminal ions
- assigned fragment ion masses stay within a coherent mass error pattern
- the precursor ion and charge state support one peptide path
- replicate scans show similar local evidence
- the b-ion series and y-ion series are sparse or broken
- non-informative peaks dominate the spectrum
- precursor isolation purity is poor
- one scan appears to contain fragments from more than one species
- strong peaks that do not form a continuous ladder
- acceptable precursor mass with weak fragment-level support
- conflicting residue assignments around one mass gap
- unresolved leucine/isoleucine ambiguity mixed with broader sequence uncertainty
- replicate scans that disagree on the same region
- ladder continuity across the N- and C-terminal directions
- fragment intensity distribution
- consistency of mass error
- signs of co-fragmentation
- dominance of neutral loss over backbone ions
- the candidate fits precursor mass and expected modification state
- consecutive ions support residue order
- both termini have at least partial evidence
- assigned fragments show coherent mass error
- replicate or orthogonal data support the same region
- a cleaner annotated MS/MS spectrum
- a more stable candidate path or sequence tag
- improved local confidence distribution across supported residues
- a clearer assessment of whether the spectrum-level evidence supports one peptide or several possibilities
- replicate LC-MS/MS acquisitions for the same precursor
- an alternate fragmentation mode when the original scan is neutral-loss dominated
- targeted validation of a proposed sequence region or modified site
- cross-checking the de novo result against precursor mass, expected PTM state, and any independent characterization data
Poor MS/MS spectrum quality makes peptide de novo sequencing break down when the fragment evidence is too weak to support residue-by-residue inference. The usual causes are discontinuous b-ion series or y-ion series, weak fragment structure relative to the signal-to-noise ratio, co-fragmentation that produces a chimeric spectrum, and post-translational modification (PTM)-driven fragmentation bias. In practice, the real question is not whether software returned a candidate. It is whether the spectrum contains enough coherent evidence to defend that candidate.
Quick Decision Guide
Use the current spectrum for cautious interpretation when:
Reacquire or redesign the workflow when:
Treat any full-sequence claim as limited when standard MS/MS leaves unresolved isobaric assignments, PTM placement, or mixed-spectrum ambiguity. A long candidate sequence is not automatically a high-confidence one.
Where This Problem Usually Appears
This pattern usually shows up after an LC-MS/MS run is finished and de novo peptide sequencing software returns short tags, multiple competing paths, or uneven local confidence. The team may already know that a standard database search is not enough because the target is novel, truncated, mutated, impurity-related, or heavily modified. The next question is whether the current tandem mass spectrometry evidence is still worth salvaging.
Typical warning signs include:
A useful de novo result depends on direct spectral evidence. Software scores can rank candidates, but they cannot replace missing fragmentation logic.
Why Poor Spectra Break De Novo Interpretation
Incomplete or discontinuous ion ladders
The clearest requirement for peptide de novo sequencing is a sequence-informative ladder. When the b-ion series or y-ion series breaks repeatedly, the algorithm cannot bridge residues with confidence. A few intense peaks may look encouraging, but isolated peaks rarely provide enough ordering information to support a defensible full-sequence call.
Weak signal-to-noise and poor fragment intensity structure
Low signal-to-noise ratio lowers confidence even when some expected fragments are present. Informative ions may sit close to chemical background, detector noise, or low-level contaminants. In that situation, the candidate path often rests on weak peaks that are easy to overread.
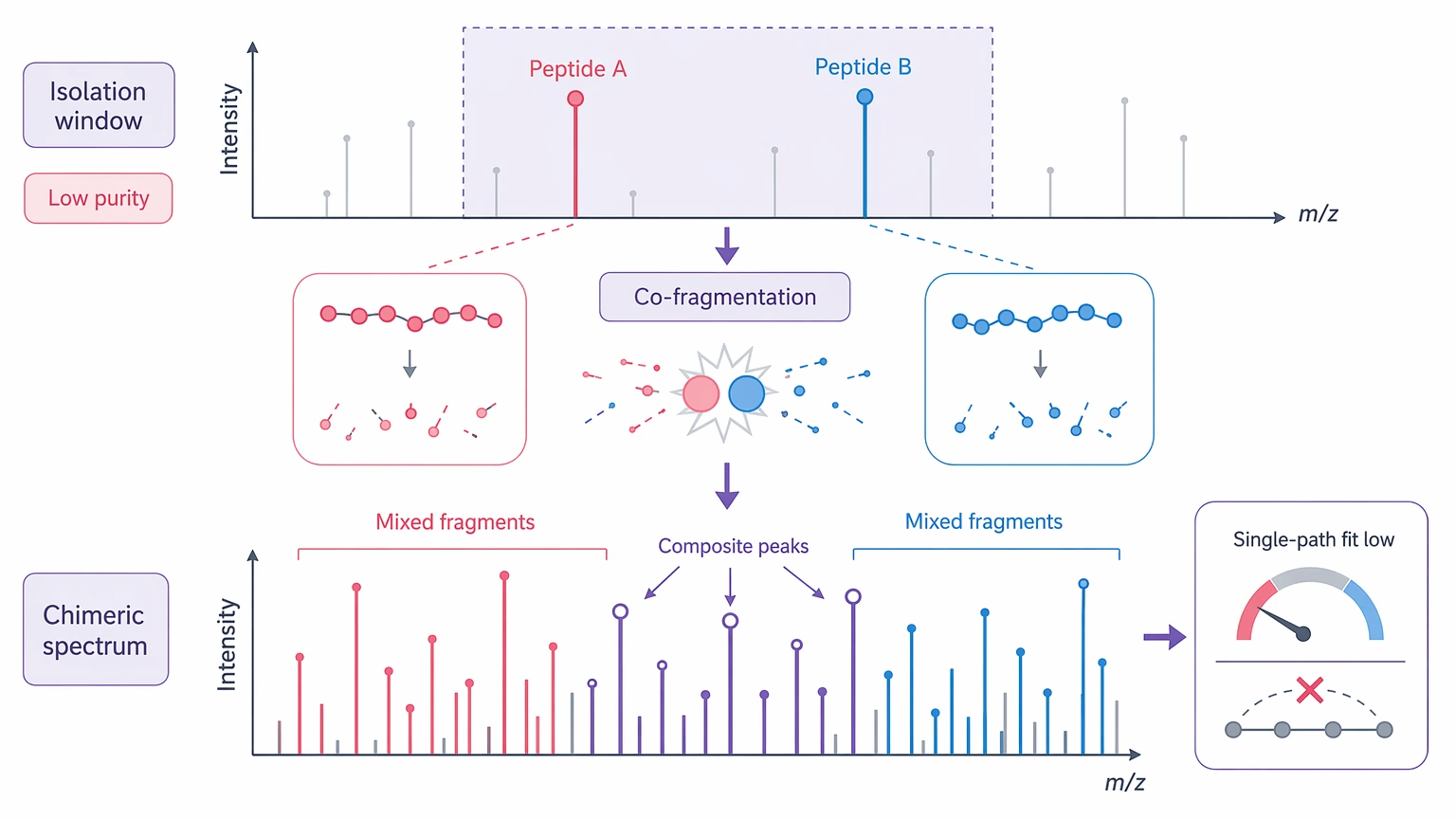
Co-isolation and chimeric spectra
Poor precursor isolation purity often leads to co-fragmentation. The resulting chimeric spectrum can look rich in fragments, but those fragments do not support a single peptide. This is one of the more deceptive failure modes because the spectrum may look information-dense while still being unusable for clean sequence reconstruction.
PTM-driven and sequence-specific fragmentation complexity
A PTM can shift expected fragment masses, favor neutral loss, and suppress backbone cleavages that de novo algorithms depend on. Some peptide backbones also fragment unevenly because of sequence chemistry, charge localization, or labile modifications. When that happens, the spectrum may support only a partial path instead of a full call.
Sample complexity that prevents clean precursor evidence
Mixed peptide populations, truncation products, adducts, salt background, or limited purification can all complicate interpretation before the spectrum is even scored. In these cases, weak sequence confidence reflects a sample-input problem as much as a software problem.
A Troubleshooting Path for Low-Confidence De Novo Calls
Step 1: Decide whether the spectrum is interpretable
Start with one precursor ion at a time. Ask whether the precursor mass, charge state, and fragment assignments fit one coherent explanation. If you cannot find even a short, internally consistent sequence tag, deeper interpretation is unlikely to rescue the call.
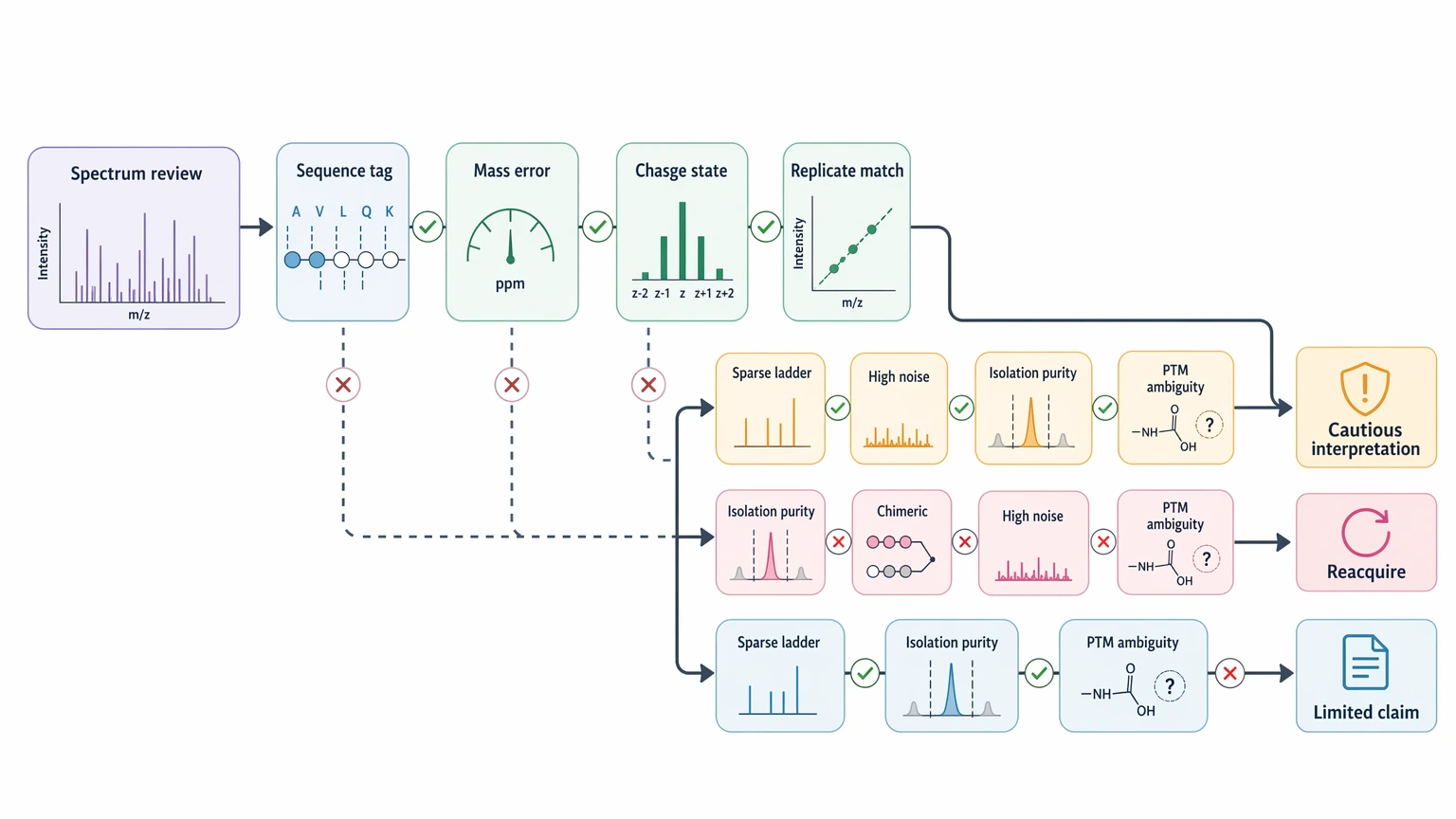
A practical first-pass screen is shown below.
| Evidence | What it supports | Main limitation | Next move |
|---|---|---|---|
| Consecutive terminal ions | Direct residue ordering | May not cover the full peptide | Extend with replicate or alternate fragmentation |
| Strong peaks without ladder continuity | Some fragmentation occurred | High risk of false path assembly | Reacquire or inspect for neutral-loss bias |
| Mixed fragment clusters | Possible multiple precursors | Candidate may be composite | Review isolation settings and chromatographic separation |
| Good precursor mass, poor fragment fit | Precursor detection is acceptable | Sequence remains unsupported | Reassess fragmentation efficiency or purity |
Takeaway: keep interpreting only when the spectrum supports one peptide path better than the alternatives.
Service Routes to Consider
For this project scenario, readers usually compare these service routes before requesting a quote or submitting samples.
Step 2: Separate recoverable defects from nonrecoverable ones
Some spectra are weak but still improvable. Others simply do not contain enough sequence logic to justify more rescoring. Review:
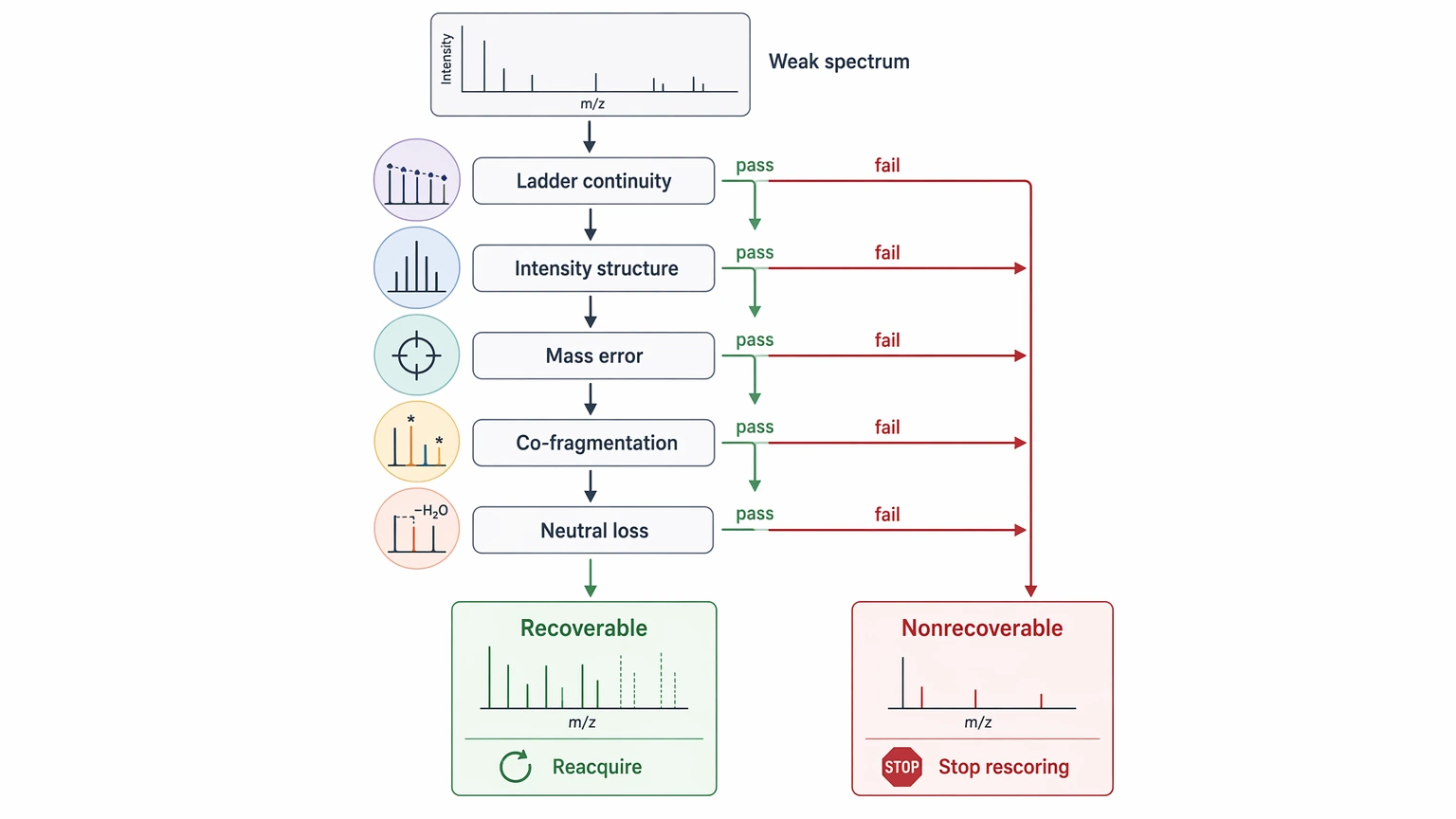
If the main gap is incomplete coverage from an otherwise workable spectrum, re-acquisition may help. If the scan is fundamentally mixed or fragment-poor, software alone will not create new evidence.
Step 3: Match the correction to the failure mode
Do not repeat the same method by default. The fix should follow the defect you actually see.
| Scenario | Recommended workflow | Key limitation | Validation need |
|---|---|---|---|
| Broken ion ladders | Optimize fragmentation conditions and precursor targeting | Some peptides still fragment unevenly | Confirm improved ladder continuity in replicate scans |
| Low signal-to-noise ratio | Clean up the sample or enrich the target if possible | Material may be limited | Compare fragment structure before and after cleanup |
| Chimeric spectrum | Tighten isolation or improve chromatographic separation | Narrower selection can reduce sensitivity | Show that one precursor yields one fragment population |
| PTM-heavy fragmentation | Use a PTM-aware interpretation plan or alternate fragmentation | Labile modifications may remain difficult | Check modification consistency across follow-up spectra |
| Complex mixed input | Simplify or purify the sample before repeat LC-MS/MS | Extra handling may consume sample | Verify cleaner precursor evidence after rework |
If your team is deciding whether to continue with re-acquisition or move to a specialist workflow, MtoZ Biolabs can evaluate your project against the current spectra, sample constraints, and reporting goal before additional material is consumed.
Step 4: Define the claim level before reporting
A partial result can still be useful. A short sequence tag may narrow an impurity hypothesis, support a modified-region assignment, or constrain later targeted work even when a full sequence remains uncertain.
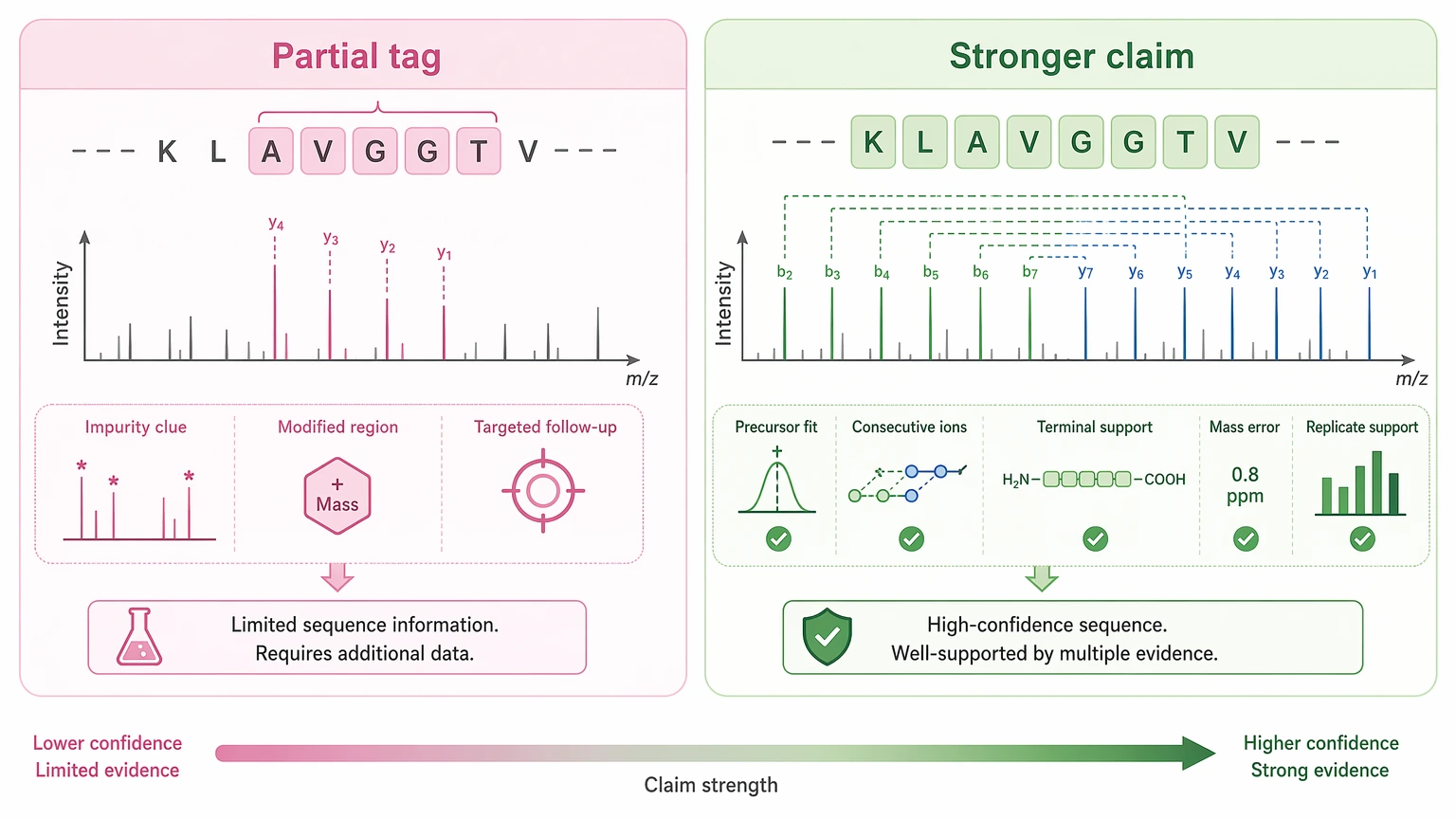
Use a stronger claim only when these points line up:
When those conditions are missing, report the output as provisional. This matters especially for protein de novo sequencing projects where downstream decisions may depend on one uncertain peptide call.
Expected Results and Validation Methods
After corrective action, the first improvement should show up in the spectrum itself, not just in the software score. You are looking for clearer terminal ladders, fewer incompatible assignments, and better agreement between precursor information and the proposed sequence.
Immediate deliverables
Follow-up confirmation
An explicit limitation matters here: standard MS/MS often cannot fully resolve leucine/isoleucine ambiguity, and PTM-rich spectra may still support more than one plausible sequence path. Database-search gaps can also remain because the best de novo candidate may represent only part of the real sequence, not the full peptide.
Key Cautions and Practical Limits
Sample amount and sample quality set real limits. Cleanup, fractionation, and repeat acquisition can improve spectrum quality, but every extra step may consume scarce material or bias what remains.
Controls and repeats matter as much as a single attractive scan. When co-fragmentation risk is high, one spectrum is rarely enough for confident reporting. Replicate evidence should confirm the same precursor behavior and the same sequence-supported region.
Batch variation and contamination can change the fragment landscape. Carryover, salts, polymer background, and low-level coeluting species can all lower sequence confidence or create false structure in the spectrum.
Interpretation also has boundaries. A candidate with good precursor agreement may still have weak residue-level support. A PTM assignment may remain site-ambiguous even when the peptide backbone is mostly correct. Put differently, spectrum-level confidence and biological certainty are not the same thing.
Another method may be the better next step when repeated runs keep showing poor isolation, unstable fragmentation, or contradictory candidates. In those cases, targeted confirmation, broader protein identification, or a different sequencing strategy may move the project forward more efficiently than another de novo attempt. If that decision point has become the main blocker, you can submit your requirements to MtoZ Biolabs for a scoped review of sample amount, acquisition history, and validation options.
Conclusion
Poor MS/MS spectrum quality causes peptide de novo sequencing to fail when the spectrum lacks continuous, clean, sequence-informative fragment evidence. The most important checks are ladder continuity, signal-to-noise ratio, precursor isolation purity, chimeric behavior, and PTM-related fragmentation complexity. Those features determine whether the data support a defensible sequence, a limited sequence tag, or a decision to stop and redesign the workflow.
This framework is most useful for unknown peptides, modified peptides, truncation products, and other database-limited projects where residue-level interpretation still matters. If your current data leave the project stuck between partial evidence and overcalling, the next practical step is to align the reporting claim with the actual spectral support, then decide whether re-acquisition, targeted validation, or outside sequencing support is the better path. For that kind of project triage, contact us with the spectra, sample context, and decision goal so the next experiment answers the real sequence question instead of repeating the same uncertainty.
FAQ
Can a weak de novo result still support impurity investigation?
Yes. Even a short sequence tag can help distinguish a sequence-related impurity from an unrelated contaminant if the tag matches the precursor mass and the suspected modification or truncation pattern.
What makes a chimeric spectrum different from an ordinary noisy scan?
A noisy scan has too much background around one peptide. A chimeric spectrum contains fragment evidence from more than one precursor, so it can create internally inconsistent residue paths even when several peaks look strong.
Should I trust a candidate with one long y-ion ladder but almost no b-ions?
Sometimes, but only cautiously. One strong terminal ladder can support part of a sequence, yet confidence drops if the opposite terminus is absent and no replicate evidence confirms the same path.
How do I tell PTM-related ambiguity from a basic sequencing error?
PTM-related ambiguity usually appears as shifted fragment clusters, repeated neutral loss, or uncertainty around modification placement. A basic sequencing error more often shows inconsistent residue differences without a convincing modification logic.
When is database search still useful after de novo analysis struggles?
Database search can still test whether the observed fragments align with truncations, expected variants, or modification-bearing candidates. It becomes especially useful when the de novo output is partial rather than fully novel.
What is a reasonable reporting format for a low-confidence call?
Use a bounded statement: report the supported sequence region, the unresolved residues or PTM positions, the main spectral limitations, and the follow-up experiment needed before making a stronger claim.
How to order?

