





Top-Down Protein Sequencing Principle: Intact Protein MS, Fragmentation, and Proteoform Analysis
-
Top-down sequencing keeps proteins intact, so modification combinations and proteoforms can be observed more directly than in bottom-up workflows.
-
The method depends on clean intact protein preparation, high-resolution MS, effective fragmentation, and specialized data analysis.
-
It is powerful for purified proteins, biopharmaceutical characterization, proteoform analysis, and PTM mapping when sequence context matters.
-
Bottom-up, middle-down, and top-down methods are complementary rather than interchangeable.
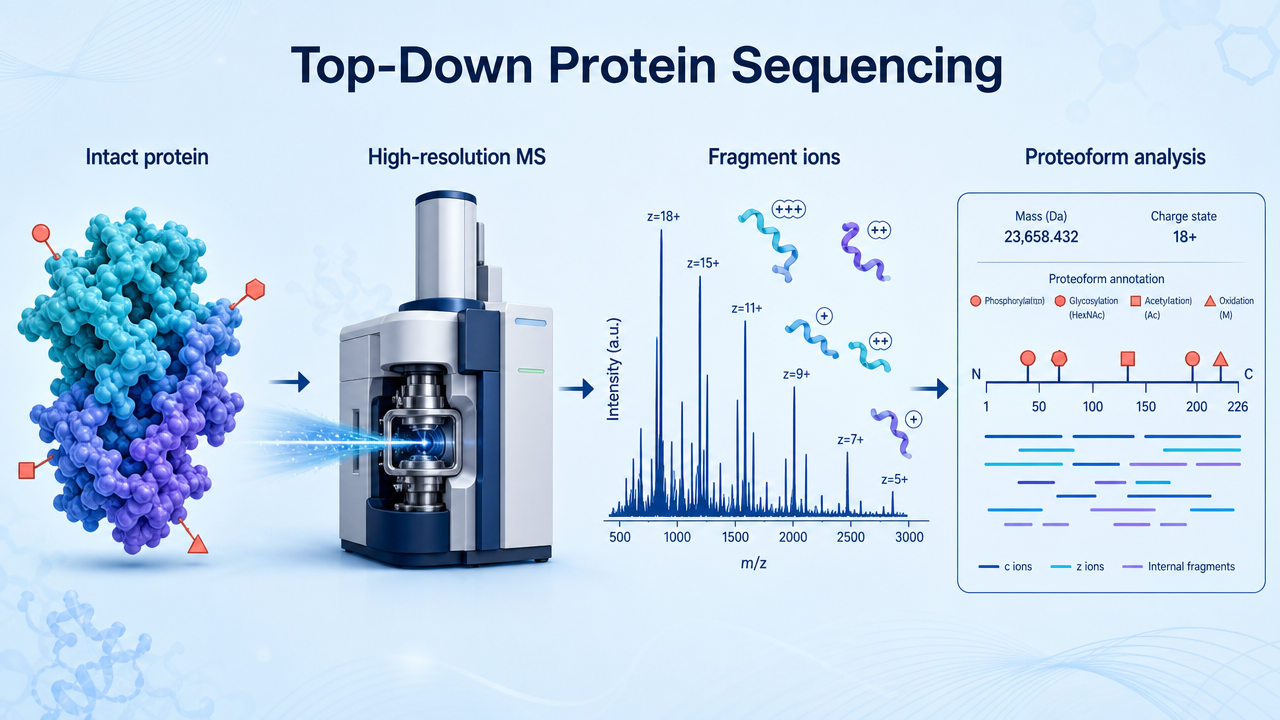
Top-down protein sequencing analyzes intact proteins directly by high-resolution mass spectrometry instead of digesting them into peptides first. The principle is simple but technically demanding: preserve the whole protein, measure its intact mass, fragment the gas-phase protein ion, and use fragment ions to infer sequence features, terminal processing, post-translational modifications, variants, and proteoforms.
Key Takeaways
What Does Top-Down Protein Sequencing Measure?
Top-down protein sequencing measures intact protein ions and their fragments. Because the protein is not first reduced to short tryptic peptides, the analysis can preserve combinations of PTMs, sequence variants, truncations, isoforms, and terminal modifications on the same molecule.
Related Services
Top-Down Protein Sequencing Service
Top-down Proteomics Analysis Service
Mass Spectrometry-Based Protein Sequencing Service
Top-Down Based PTMs Characterization Service
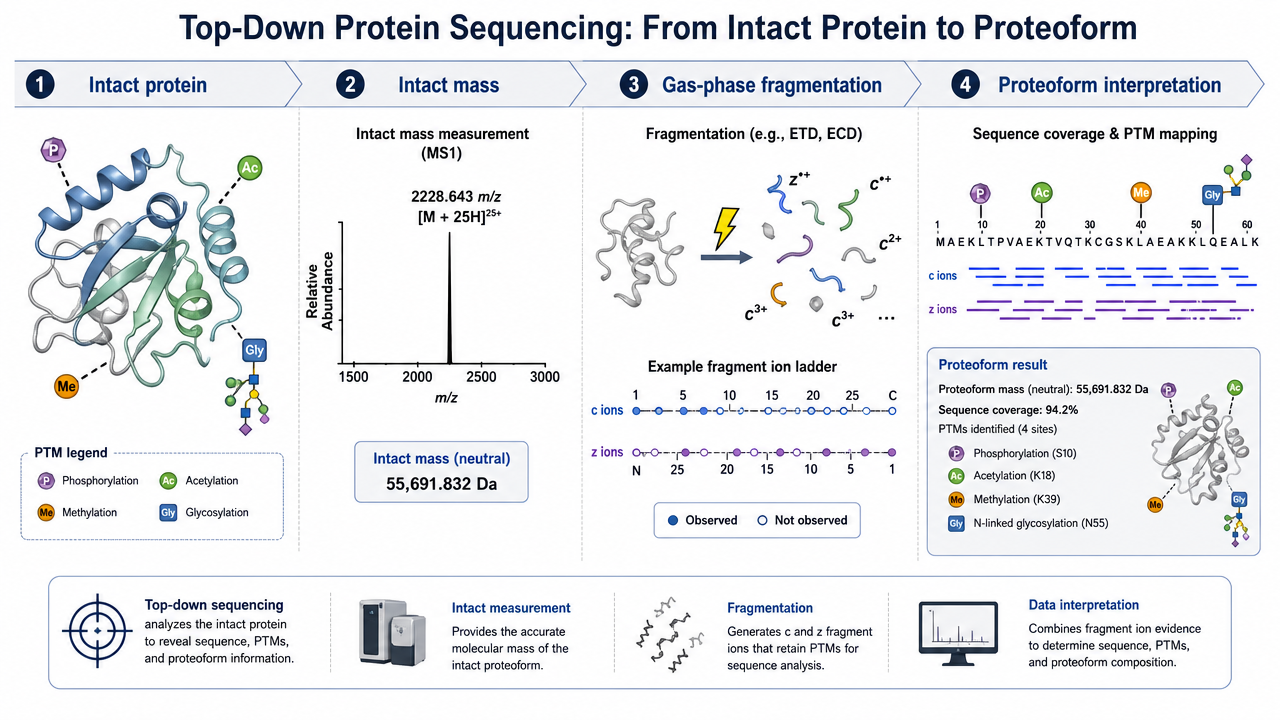
Step 1: Preserve and Prepare Intact Proteins
Sample preparation is the first constraint. Salts, detergents, polymers, and buffer components can suppress ionization or broaden peaks. Proteins may need purification by affinity chromatography, ion exchange, size exclusion, HPLC, gel-based separation, buffer exchange, or desalting before MS.
Step 2: Measure Intact Protein Mass
The intact protein is ionized and measured by high-resolution MS. Instruments such as Orbitrap or FT-ICR platforms can resolve charge-state envelopes and provide accurate molecular weight information. This intact mass already gives useful information: truncation, oxidation, glycosylation, adducts, disulfide state, or unexpected proteoforms may shift the observed mass.
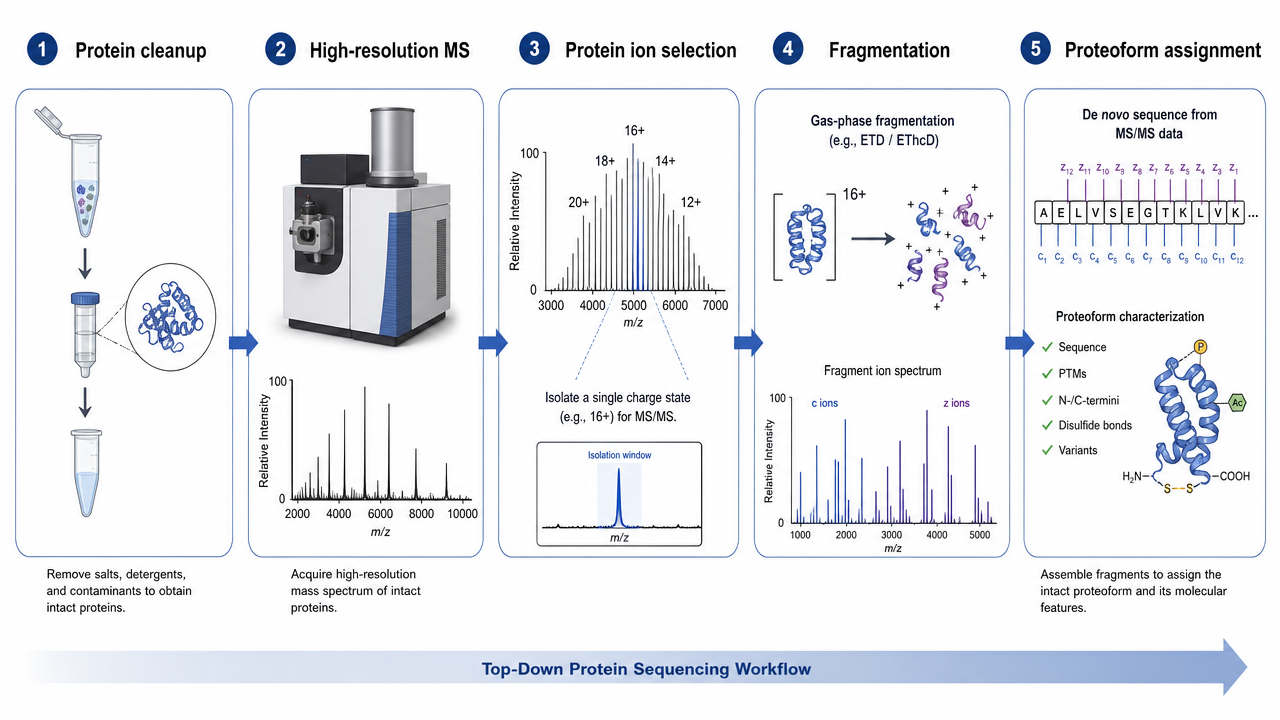
Step 3: Fragment Intact Protein Ions
After intact mass measurement, selected protein ions are fragmented in the gas phase. Fragmentation methods may include electron-based dissociation or collision-based approaches, depending on instrument configuration and protein properties. The goal is to generate sequence-informative fragment ions while preserving enough modification information to localize changes.
Step 4: Interpret Proteoforms and PTMs
Top-down data analysis matches intact mass and fragment ions against protein sequences. The output may include sequence confirmation, proteoform assignment, terminal processing, truncation, variant detection, and PTM localization. The key advantage is proteoform context: modification combinations can sometimes be preserved on the same protein molecule.
Main Applications
Top-down protein sequencing is useful for biopharmaceutical characterization, intact protein confirmation, proteoform analysis, PTM characterization, protein variant detection, terminal processing studies, and purified protein QC.
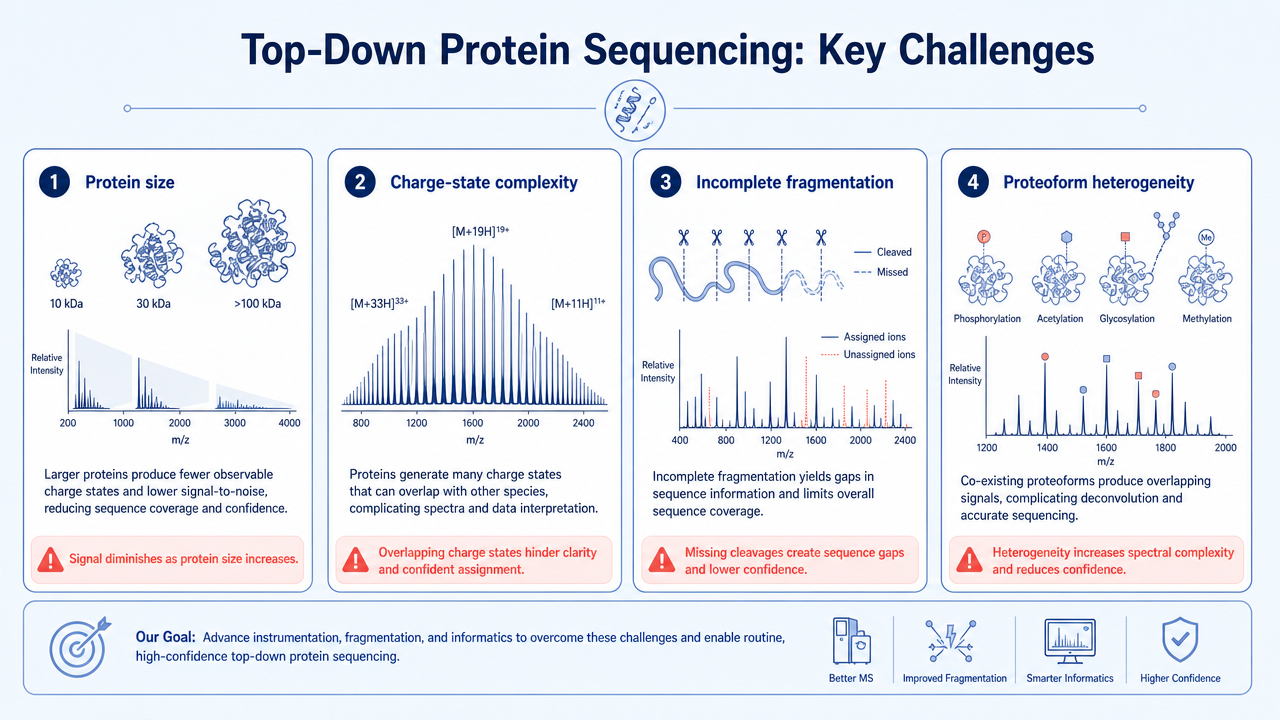
Main Limitations
Top-down protein sequencing is technically demanding. Large proteins are harder to ionize, fragment, and interpret. Mixtures create overlapping charge envelopes. PTM heterogeneity can split signal across many proteoforms. Data analysis requires careful mass accuracy, fragment matching, and false-discovery control.
Top-Down Vs Bottom-Up Vs Middle-Down
| Approach | Starting Material | Best for | Main Tradeoff |
|---|---|---|---|
| Bottom-up | Digested peptides | Broad proteome coverage and routine identification | Loses some proteoform context |
| Middle-down | Large peptides or protein domains | PTM combinations over longer sequence regions | Requires tailored digestion and analysis |
| Top-down | Intact proteins | Proteoforms, variants, intact mass, PTM combinations | Harder for complex mixtures and large proteins |
FAQ
1. What is the principle of top-down protein sequencing?
Top-down protein sequencing measures intact protein ions, fragments them in the mass spectrometer, and interprets fragment ions to identify sequence features, variants, PTMs, and proteoforms.
2. How is top-down different from bottom-up proteomics?
Bottom-up proteomics digests proteins into peptides before MS analysis. Top-down proteomics analyzes intact proteins first, preserving more proteoform-level context.
3. Why is top-down useful for PTM analysis?
Top-down analysis can preserve combinations of PTMs on the same protein molecule, which can be difficult to reconstruct from short peptides.
4. What samples work best for top-down sequencing?
Purified proteins, biopharmaceutical proteins, enriched protein fractions, and relatively simple mixtures are usually better suited than highly complex lysates.
Conclusion
Top-down protein sequencing is valuable when intact protein context matters. It can connect mass, sequence, PTMs, variants, and proteoforms in ways that peptide-first workflows may miss. The method works best when sample cleanup, high-resolution MS, fragmentation strategy, and data interpretation are designed around the intact protein question.
How to order?

