





SILAC Proteomics Service
SILAC proteomics is a mass spectrometry-based technique that uses stable isotope-labeled amino acids to quantify protein abundance, enabling comparative analysis of protein expression in different samples. SILAC is based on direct addition of selected stable isotope amino acids into the cell culture medium, allowing superior quantitative proteomics compared to other labeling methods. The great advantages of SILAC lie in its straight-forward implementation, quantitative accuracy, and reproducibility over chemical labeling or label-free quantification strategies, favoring its adoption for proteomic research. SILAC proteomics has been widely applied to characterize the proteomic changes between different biological samples, to investigate dynamic changes of protein PTMs, to distinguish specific interacting proteins in interaction proteomic analysis, and to analyze protein turnover in the proteome-wide scale, making it an important tool in functional proteomics research to answer important questions in diverse areas of biomedical research.
1. The Principal of SILAC
The principle of SILAC (Stable Isotope Labeling by Amino acids in Cell Culture) is based on metabolically incorporating stable isotope-labeled amino acids (such as 13C or 15N-labeled arginine or lysine) into the entire proteome of cells during protein metabolism, particularly during cell culture. SILAC proteomics involves culturing two populations of cells in different media, where the "light" medium contains naturally occurring isotopic amino acids and the "heavy" medium contains stable isotope-labeled amino acids. After a sufficient number of cell divisions (at least five cycles in mammalian cells), theoretically, all proteins from the cells grown in the heavy medium should contain stable isotope-labeled amino acids. However, the number of cell divisions required for complete labeling depends on the rates of protein synthesis, degradation, and turnover, so labeling efficiency must be carefully tested before quantification. Once labeling is complete (with at least 95% labeling efficiency), the cell populations are experimentally manipulated, and equal amounts of labeled and unlabeled cells or protein extracts are mixed. The samples are then digested into peptides and analyzed using liquid chromatography-tandem mass spectrometry (LC-MS/MS). The quantification of SILAC is based on measuring the ratio of isotope-labeled peptides to unlabeled peptides, allowing for the quantitative comparison of their relative abundances in the mixture by comparing the signal intensities of the light and heavy samples.
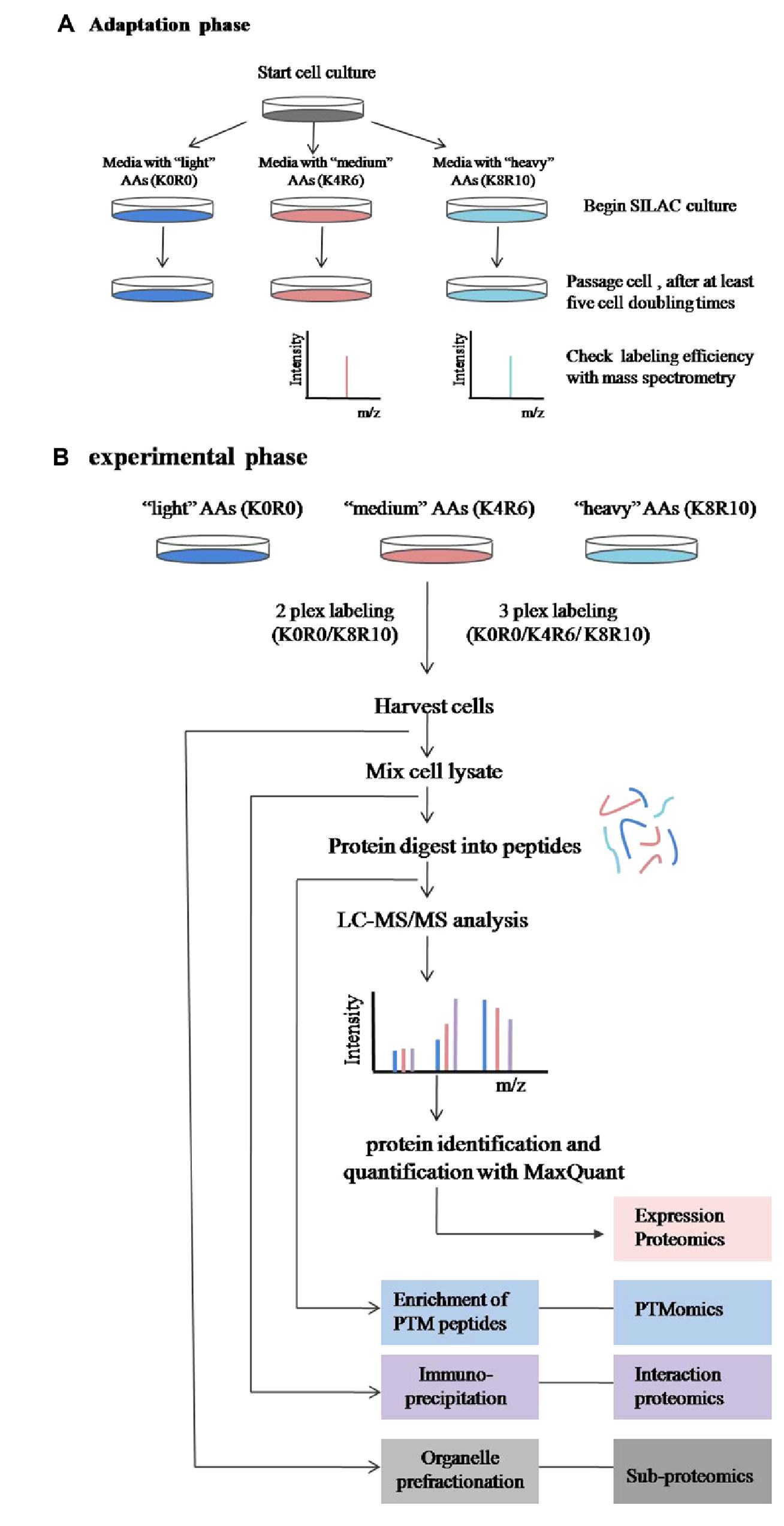
Chen, X. et al. Proteomics. 2015.
2. Advantage of SILAC
2.1 Quantification Challenges in MS and Solution through Stable Isotope Labeling
Quantitative proteomics based on mass spectrometry (MS) is a powerful tool in biological research. However, MS itself is inherently non-quantitative because peptides, due to their different physicochemical properties (such as size, charge, and hydrophobicity), have different responses in the mass spectrometer. This makes it difficult to compare relative abundance changes between different experimental samples in the same MS analysis. To address this issue, SILAC proteomics is introduced to enable relative quantification of proteins from different samples within the same MS analysis. This approach reduces variability caused by sample injection and ion suppression, improving the accuracy of the results.
2.2 Advantages of SILAC Compared to Label-Free Methods
In contrast to label-free methods, which do not require chemical labeling of proteins or peptides, SILAC proteomics uses stable isotope-labeled amino acids for quantification. This allows for the direct combination of differentially treated samples at the protein or cellular level and minimizes experimental error or bias. Label-free methods typically rely on measuring ion intensity variations or spectral counting to compare samples, but because no labeling is involved, they can suffer from lower precision. SILAC proteomics provides more accurate quantitative data, particularly in complex biological samples, as it enables precise quantification of changes in protein abundance.
2.3 Expansion of SILAC Applications
The development of SILAC proteomics has expanded its applications beyond cell culture proteomics. With advancements such as isotope-labeled SILAC and super SILAC, this technique has been extended to tissue samples and body fluids, enabling large-scale proteomic analysis in more complex biological systems.
3. Applications of SILAC
3.1 Expression Proteomics
Organoids have the potential to link 3D cell culture with tissue physiology by providing models that resemble in vivo organs. The technology began with the isolation of long-term growing organoids from intestinal crypt cells, which led to the reconstruction of the renewed intestinal epithelial niche. Since then, this breakthrough has been applied to many other organs, including the prostate, liver, kidney, and pancreas. Researchers described how to apply SILAC-based quantitative proteomics to measure changes in protein expression in intestinal organoids under different experimental conditions. They developed SILAC organoid culture media that allowed the organoids to grow and differentiate normally, and confirmed the incorporation of isotope-labeled amino acids. Additionally, the researchers used a treatment known to affect organoid differentiation to demonstrate the reproducibility of the method for quantitative analysis and to validate the identification of proteins associated with the inhibition of cell growth and development. By combining quantitative mass spectrometry, SILAC, and organoid culture, the researchers validated the method and showed that large-scale proteomic variation can be measured in "organ-like" systems.
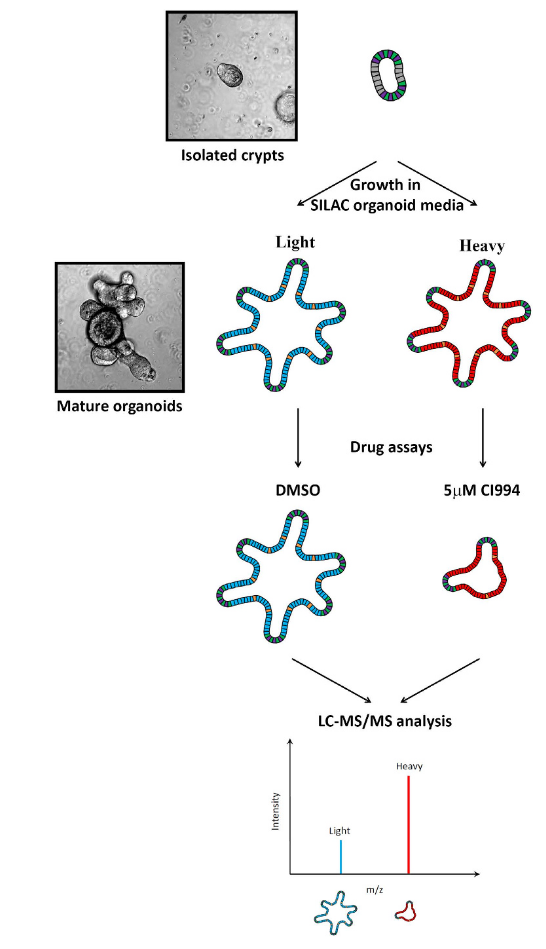
Gonneaud, A. et al. Sci Rep. 2016.
3.2 Dynamic Changes of Protein PTMs
Post-translational modifications (PTMs) of proteins, particularly acetylation, phosphorylation, and ubiquitination, play key roles in the host's innate immune response. The dynamic changes in PTMs and their crosstalk are complex. To construct a comprehensive dynamic network of inflammation-related proteins, researchers integrated data from the whole-cell proteome (WCP), acetylome, phosphoproteome, and ubiquitome of human and mouse macrophages. Their datasets on acetylation, phosphorylation, and ubiquitination sites contribute to identifying the crosstalk between PTMs within and between proteins involved in the inflammatory response. Lipopolysaccharide (LPS) stimulation of macrophages leads to both degradative and non-degradative ubiquitination. Additionally, integrative SIlAC proteomics helps to elucidate the roles of known inflammatory molecules and discover novel inflammatory proteins.
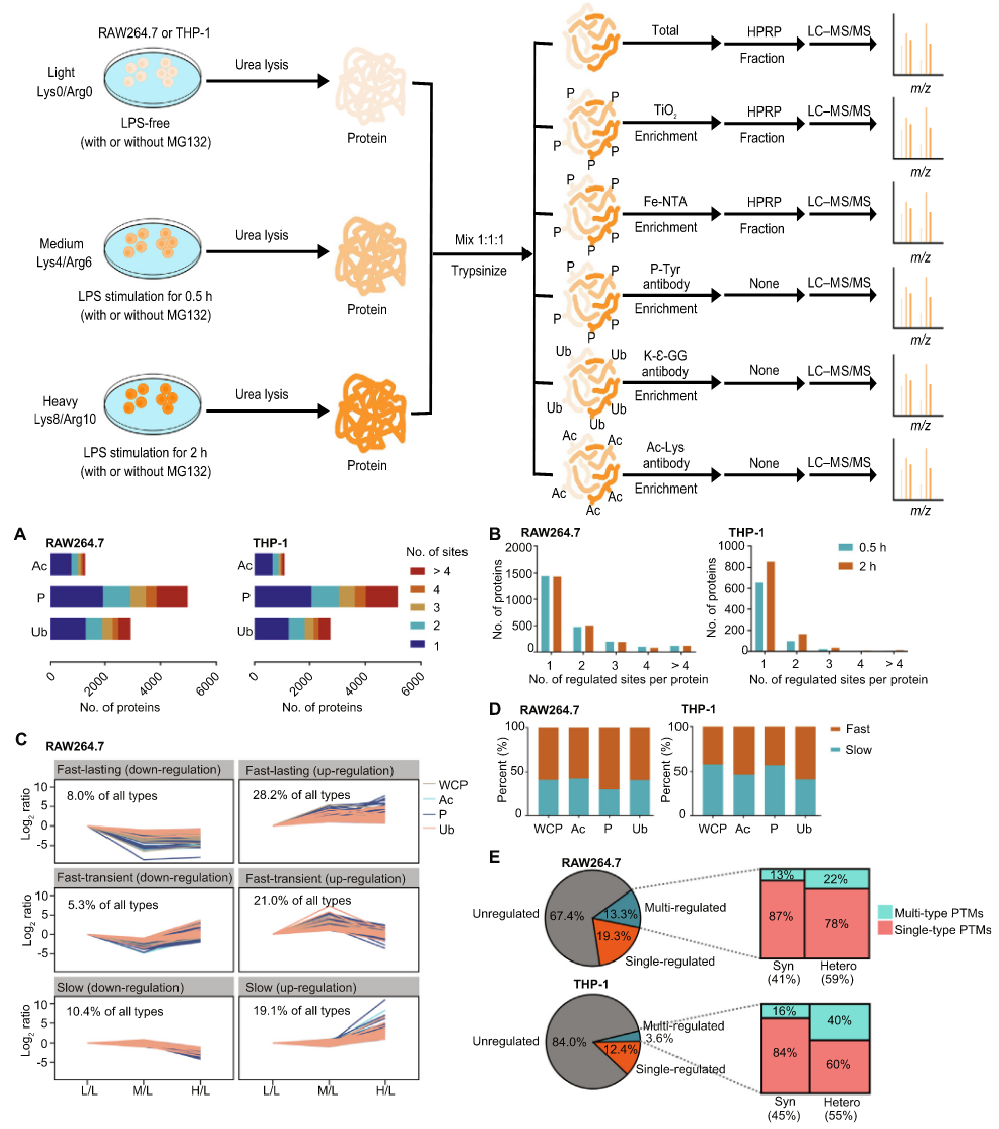
Ji, F. et al. Genomics Proteomics Bioinformatics. 2022.
3.3 Interaction Proteomics (Interactomics)
Progesterone receptor (PR) isoforms PRA and PRB exert their effects in both progesterone-independent and progesterone-dependent manners, differentially regulating the biological characteristics of breast cancer cells. Here, we demonstrate that structural differences between PRA and PRB promote the binding of both common and distinct protein interaction partners, thereby influencing downstream signaling events for each PR isoform. Tet-inducible HA-tagged PRA or HA-tagged PRB constructs were expressed in T47DC42 (PR/ER-negative) breast cancer cells. Affinity purification combined with stable isotope labeling by amino acids in cell culture (SILAC) mass spectrometry was used to comprehensively study the interaction partners of PRA and PRB under both ligand-free and ligand-bound conditions. To validate our findings, we applied forward and reverse SILAC conditions to effectively reduce experimental errors. These datasets will aid in the study of PRA and PRB-specific molecular mechanisms and serve as a database for subsequent experiments to identify novel PRA and PRB interaction proteins that mediate differential biological functions in breast cancer.
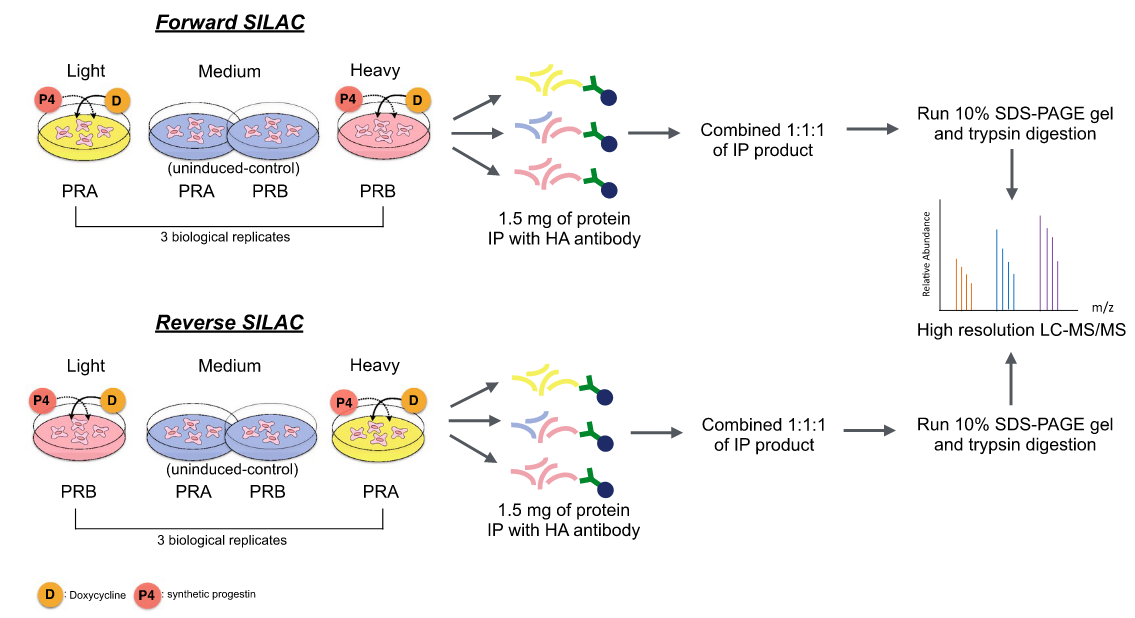
Pateetin, P. et al. Sci Data. 2021.
3.4 Protein Turnover
The balance between protein synthesis and degradation is known as protein turnover, which is crucial for cellular protein homeostasis. However, a SILAC proteomic analysis of protein turnover in adipocytes has not been conducted. Adipocytes are well-known for their role in energy storage and their association with obesity and metabolic disorders. With this objective in mind, a study employed time-dependent SILAC labeling to assess protein turnover in 3T3-L1 adipocytes over a period of 0 to 144 hours. The researchers observed relatively fast or slow protein half-lives in several proteomes, which were associated with the PPARγ signaling pathway, energy metabolism, extracellular matrix, ubiquitin-proteasome system, RNA splicing, Golgi complex, and lysosomes. These protein half-life profiles are expected to provide a clearer understanding of the adipocyte proteome lifecycle and shed light on how adipocytes maintain protein homeostasis.
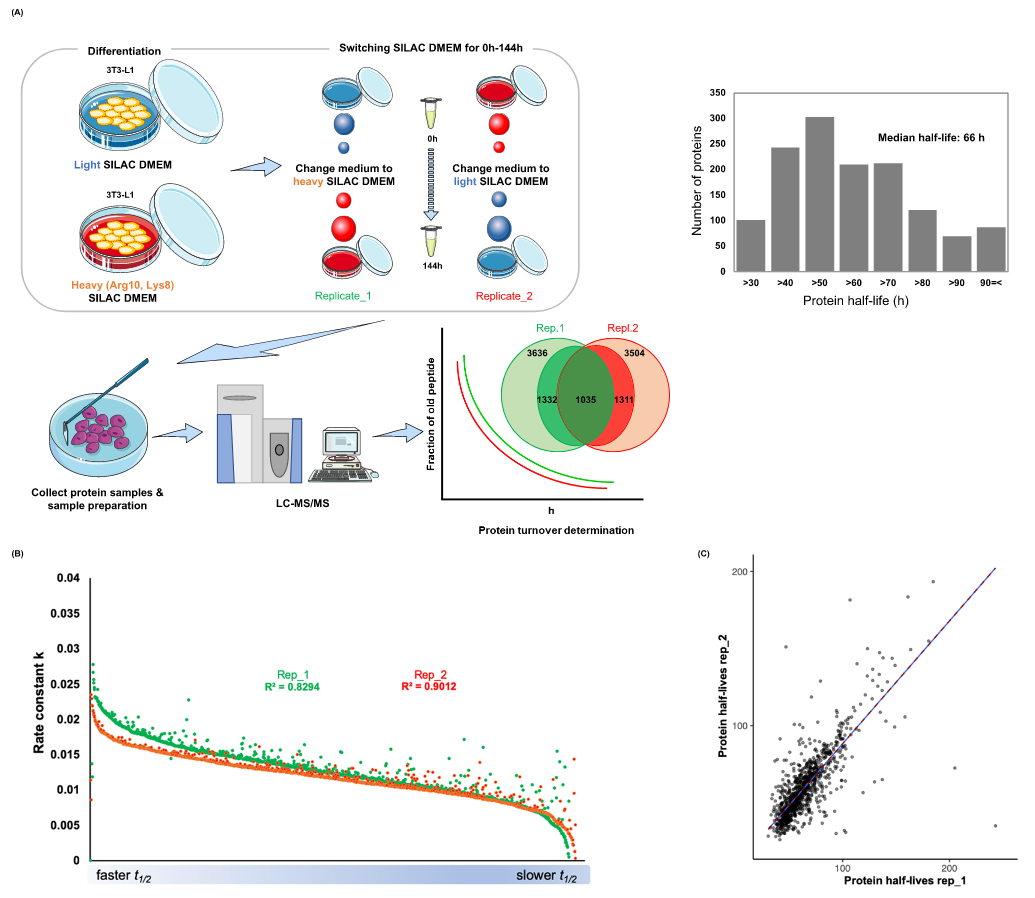
Choi, S. et al. J Proteome Res. 2023.
Service at MtoZ Biolabs
MtoZ Biolabs, an integrated chromatography and mass spectrometry (MS) services provider, provides advanced proteomics, metabolomics, and biopharmaceutical analysis services to researchers in biochemistry, biotechnology, and biopharmaceutical fields. Our ultimate aim is to provide more rapid, high-throughput, and cost-effective analysis, with exceptional data quality and minimal sample consumption. We are honored to introduce our SILAC proteomics services equipped with the Thermo Fisher Q Exactive HF and Orbitrap Fusion Lumos mass spectrometer systems, coupled with a Nano-LC system. We have extensive experience in SILAC proteomics and are continuously improving our methods, receiving much praise from our clients. We guarantee to provide professional services that meet the diverse needs of our customers. If you are interested in our services, please feel free to contact us.
Service Advantages
1. Precise Quantitative Analysis
MtoZ Biolabs leverages SILAC technology to provide customers with high-precision protein quantification services. The company ensures that each experiment accurately measures changes in protein abundance through expert mass spectrometry analysis and advanced data processing techniques, helping clients obtain reliable experimental results.
2. Optimized Experimental Workflow
MtoZ Biolabs simplifies the experimental design and optimizes operational workflows in SILAC proteomics services. With its extensive experience, the company ensures that clients can efficiently perform cell labeling and subsequent analysis, avoiding operational errors common in traditional methods, thus improving experimental efficiency and accuracy.
3. Wide Application Support
MtoZ Biolabs provides SILAC proteomics services for a wide range of cell lines based on client needs. The company’s flexible service offerings ensure that clients with diverse research requirements can successfully implement high-quality quantitative analysis, regardless of cell type.
Deliverables
1. Comprehensive Experimental Details
2. Materials, Instruments, and Methods
3. Relevant Liquid Chromatography and Mass Spectrometry Parameters
4. The Detailed Information of SILAC Proteomics
5. Mass Spectrometry Image
6. Raw Data
MtoZ Biolabs, an integrated chromatography and mass spectrometry (MS) services provider.
Related Services
SILAC Based Co-IP-MS for Protein Interaction Analysis Service
How to order?

