





Protein Sequencing Technology: Principles, Methods and Applications
- It is only applicable to purified samples with a free N-terminal.
- The sequence length is limited, typically not exceeding 30-50 residues.
- It cannot handle mixtures or modified proteins.
- Loss of sequence context information, which affects the reconstruction of modifications across peptide fragments.
- Modification localization depends on fragment completeness, leading to potential identification ambiguities.
- For highly heterogeneous proteins, fusion proteins, or non-model organism proteins, de novo sequencing still faces accuracy bottlenecks.
- Post-translational modification (PTM) identification
- Distinguishing isoforms based on isoelectric points
- Biopharmaceutical quality attribute evaluation
- Difficulty in interpreting the fragmentation patterns of high molecular weight proteins
- Extremely high demands on the resolution and ion accumulation capabilities of the mass spectrometry platform
- Strict control over sample purity and stability
- Representative platform: Oxford Nanopore
- Technological advantages: In theory, it can provide linear sequence information without disrupting the protein structure.
- Representative platforms: Quantum-Si, Alamar Biosciences
- Technological advantages: In theory, this method can be applied to trace amounts of samples in complex backgrounds and supports highly specific identification of certain amino acids.
In modern life sciences, the primary structure of a protein, its amino acid sequence, is no longer seen as a static molecular code. It not only dictates the protein's three-dimensional structure and functional properties but also profoundly impacts cellular behavior, signaling pathways, and even evolutionary paths and disease mechanisms. As a result, obtaining an accurate protein sequence is the starting point for a deeper understanding of its biological function and mechanisms of action.
Unlike traditional "protein identification", which focuses on "classification and recognition", protein sequencing centers on obtaining the complete and continuous amino acid information needed to analyze unknown proteins, identify sequence mutations, verify drug protein consistency, or track post-translational modifications. In fields requiring high precision, such as biopharmaceutical development, personalized medicine, structural biology, and antibody engineering, the refined capabilities of protein sequencing technology have become a critical lever in research and application decisions.
Select Service
Protein Sequencing Service by Edman Degradation
Protein Sequencing Service by Mass Spectrometry
Protein Full-Length Sequencing Service
Unknown Proteins Sequencing Service
Protein De Novo Sequencing Service
Top-Down Protein Sequencing Service
Single-Molecule Protein Sequencing Service
Protein Sequencing ≠ Protein Identification
In proteomics research, terms like "sequencing" and "identification" are often used interchangeably, leading to misapplication of terms, incorrect technical choices, and even confusion in research hypotheses.
By definition, protein sequencing refers to the process of analyzing the amino acid sequence of a protein, with the aim of reconstructing its primary structure in a continuous arrangement, typically represented from the N-terminus to the C-terminus. The goal is to obtain the "sequence itself", rather than simply determining the protein's "existence" or "classification".
In contrast, Protein Identification: Relies on mass spectrometry and database matching to determine whether a peptide "belongs" to a known protein. It is a categorical "assignment" process, not a point-by-point reconstruction of information.
Protein Sequencing Technology
1. Edman Degradation: The Beginning of High-Precision Linear Recognition
Edman degradation, developed by Pehr Edman in the 1950s, was the first chemical method for protein sequence analysis. This method relies on the specific reaction of phenylisothiocyanate (PITC) with the N-terminal amino acid. Through sequential labeling and cleavage, one PTH-amino acid is released and identified in each cycle, enabling the reconstruction of the linear sequence.
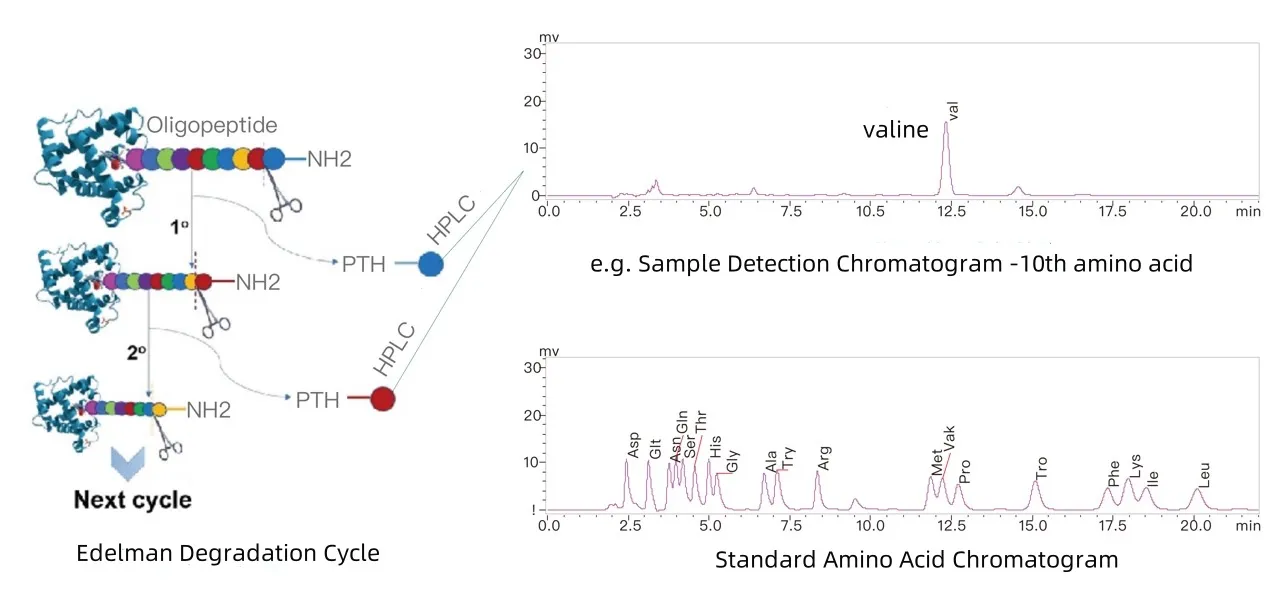
Schematic Diagram of Edman Degradation Method
The key advantages of the Edman method include: Extremely high precision at the single amino acid level, with controllable errors. However, the method also has notable limitations:
Despite its drawbacks, Edman degradation remains uniquely advantageous in areas such as validating synthetic peptide quality, evaluating recombinant protein N-terminal signal peptide cleavage efficiency, and assessing the structural consistency of biopharmaceuticals.
2. Bottom-up Strategy: The Core of High-Throughput Sequencing Technology
The introduction of mass spectrometry marked a pivotal turning point in the development of protein sequencing. The Bottom-up strategy involves digesting proteins into short peptide fragments using specific proteases (e.g., trypsin), followed by fragmentation and detection of these peptides using LC-MS/MS. The protein sequence is then reconstructed through database matching or de novo algorithms.
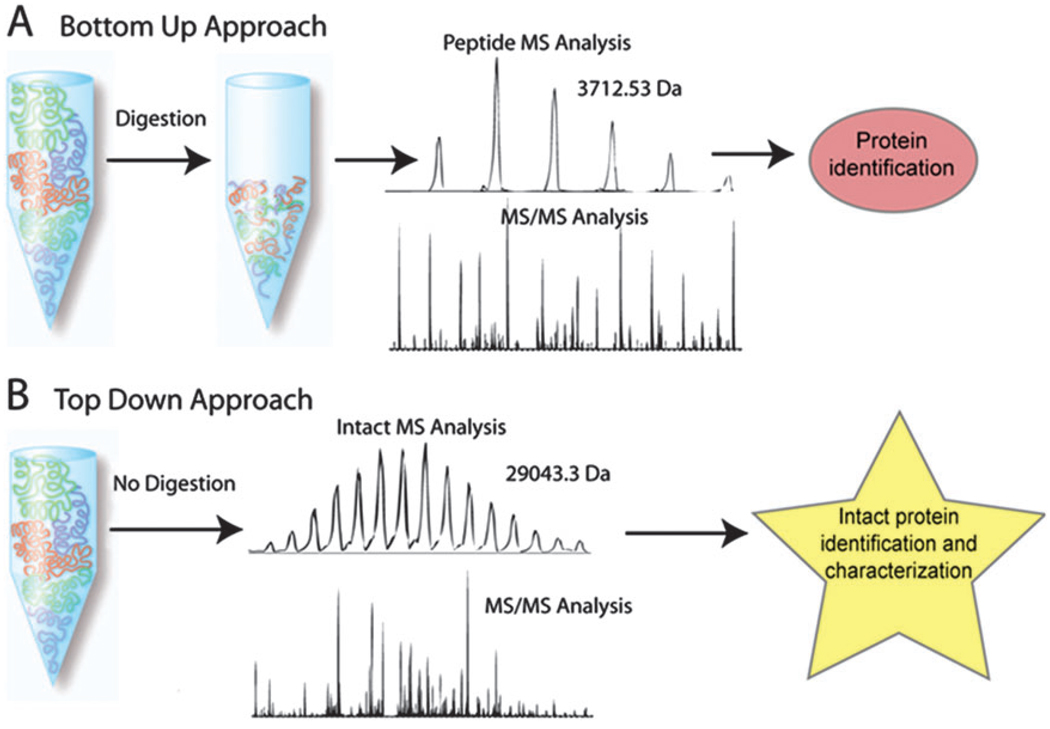
Kellie, J. F. et al. Mol Biosyst. 2010.
Schematic of the Bottom Up and Top Down approaches to protein identification
This strategy offers excellent throughput, coverage, and adaptability, making it the most widely used protein sequencing method today. However, the Bottom-up approach is inherently an indirect sequence inference method, and its sequencing capabilities are subject to the following structural limitations:
Nevertheless, the Bottom-up strategy remains an irreplaceable core tool in fields such as proteomics, antibody sequence screening, and biopharmaceutical residue analysis.
3. Top-down Strategy: In-Depth Expansion of Native Protein Structure Recognition
The Top-down sequencing strategy bypasses the digestion step and directly analyzes intact proteins by sending them into high-resolution mass spectrometry systems. High-energy fragmentation techniques, such as ETD, HCD, and EThcD, are used to generate multi-stage fragment ions, allowing for the analysis of the protein’s primary structure at the molecular level.
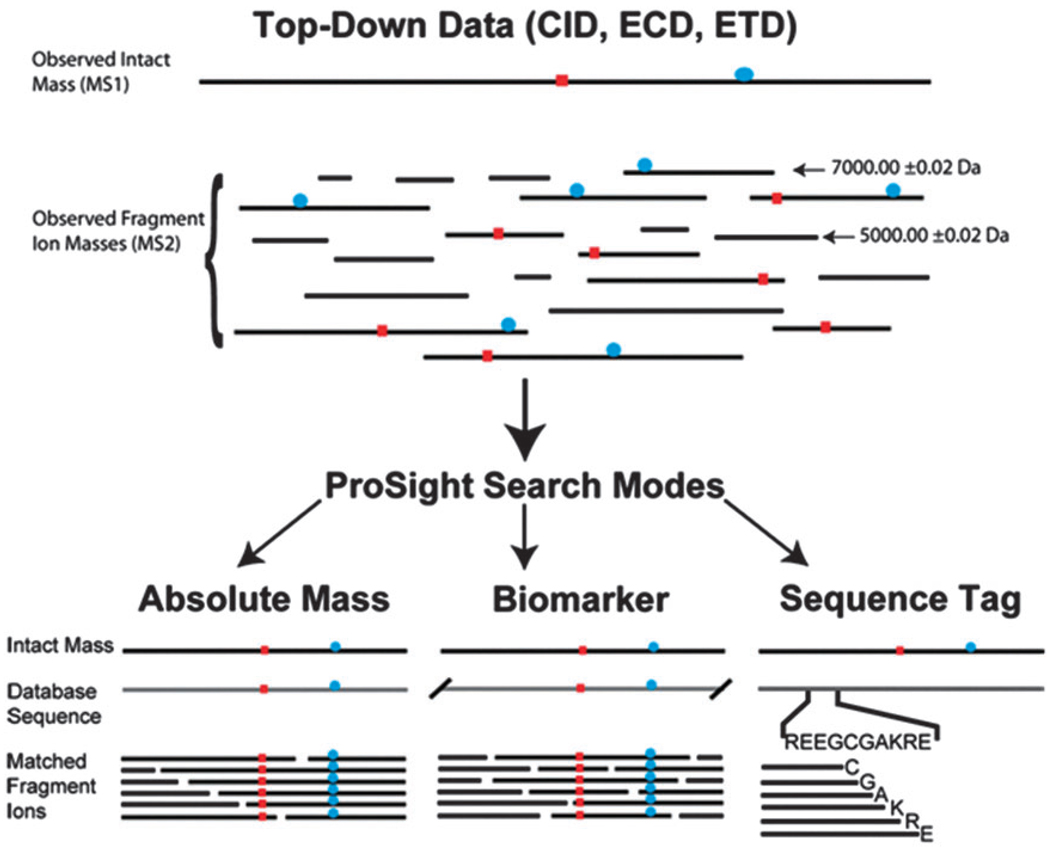
Kellie, J. F. et al. Mol Biosyst. 2010.
Schematic of the Top Down identification process
This method provides precise sequence and modification localization information without disrupting the protein’s natural modification landscape, making it particularly suitable for:
The main technical challenges of Top-down include:
While Top-down may not match Bottom-up in terms of throughput and sample adaptability, it remains irreplaceable in certain research directions, especially where structural continuity, modification fidelity, and clear isoform separation are critical. Applications include micro-mutation identification in antibody CDR regions, constructing PTM interaction networks, and ensuring structural consistency in biopharmaceuticals.
4. The Rise of Single-Molecule Protein Sequencing
Traditional mass spectrometry sequencing strategies are based on the logic of peptide fragmentation and spectrum inversion to reconstruct the sequence. While these methods are well-established, high-throughput, and highly adaptable, they inherently rely on fragment information, which introduces limitations such as resolution ambiguity, loss of modifications, and missing intra-chain associations.
In recent years, researchers have begun developing single-molecule protein sequencing (SMPS) technologies, which do not require digestion or sequence assembly and can directly read the amino acid sequence. The core idea of these technologies is to enable residue-by-residue identification of a single molecule without disrupting its overall structure.
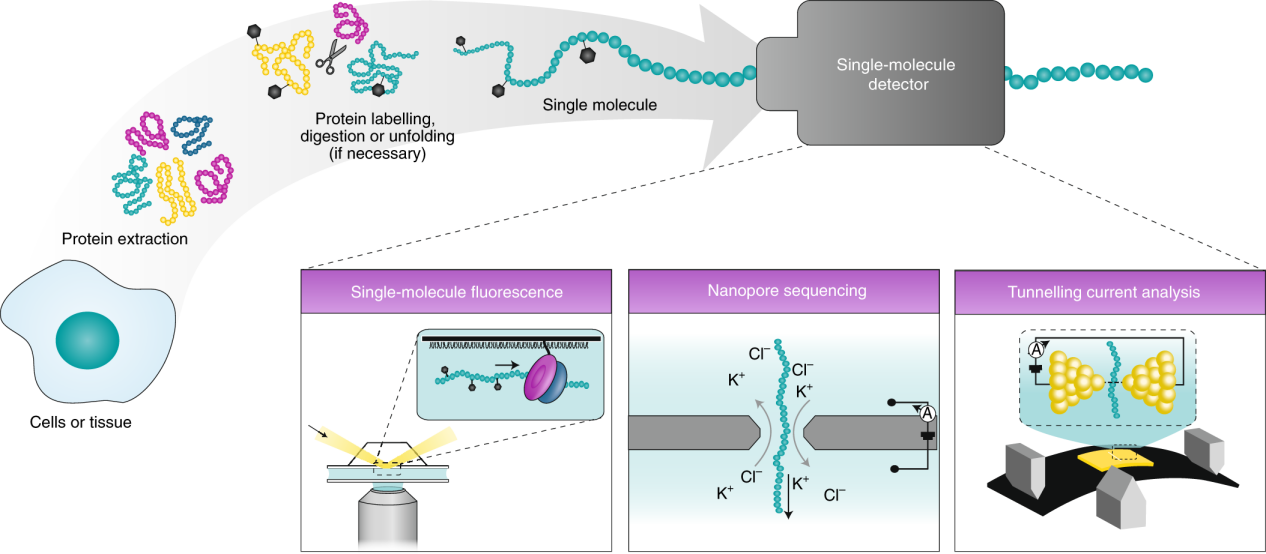
Restrepo-Pérez, L. et al. Nature Nanotech. 2018.
Schematic Of The Single-Molecule Protein Sequencing Workflow
(1) Nanopore Sequencing:
Nanopore sequencing relies on the sub-nanometer spatial resolution of biological or solid-state nanopores. Protein or short peptide molecules are guided through the pore, where the ionic current blockade caused by the molecule's passage is monitored. The properties of different amino acid residues, such as their volume, charge, and hydrophobicity, influence their spatial occupation within the pore and the local current disturbance, producing distinctive signals.
(2) Fluorescence-Based Sequencing
Fluorescence sequencing methods involve introducing identifiable fluorescent tags onto specific amino acid types and immobilizing the peptide chain onto a surface carrier. Sequential chemical treatments or enzymatic degradation are then applied to progressively remove residues, while a highly sensitive multi-channel fluorescence imaging system records the spectral characteristics of the signal released during each cycle, thereby deducing the residue sequence.
Protein sequencing lacks template dependence, exhibits inherent heterogeneity, and often involves multiple post-translational modifications. As a result, sequencing results typically do not have a singular outcome. Instead, they are based on a dynamic trade-off between experimental strategies, physical resolution, and data models. The sequencing techniques reviewed here, from chemical degradation to mass spectrometry and direct single-molecule reading, form a compensatory information acquisition system. These methods are not mutually exclusive but offer strategic support for balancing structural accuracy, sequence coverage, and modification fidelity under different research goals and sample constraints.
How to order?

