





PhIP-Seq Workflow Explained: Library Design, Sample Incubation, Sequencing, QC, and Data Review
- Peptide Array-Based Epitope Mapping Service
- Antibody Epitope Mapping Service
- Protein Array Analysis Service
A PhIP-Seq project works best when it is planned as a chain of decisions: define the antigen space, select or design the phage-displayed peptide library, plan samples and controls, run antibody-peptide binding and immunoprecipitation, sequence enriched phage DNA, review QC, then rank enriched peptides for validation. One rule matters early: agree on library design, controls, sequencing, and data review criteria before samples are committed.
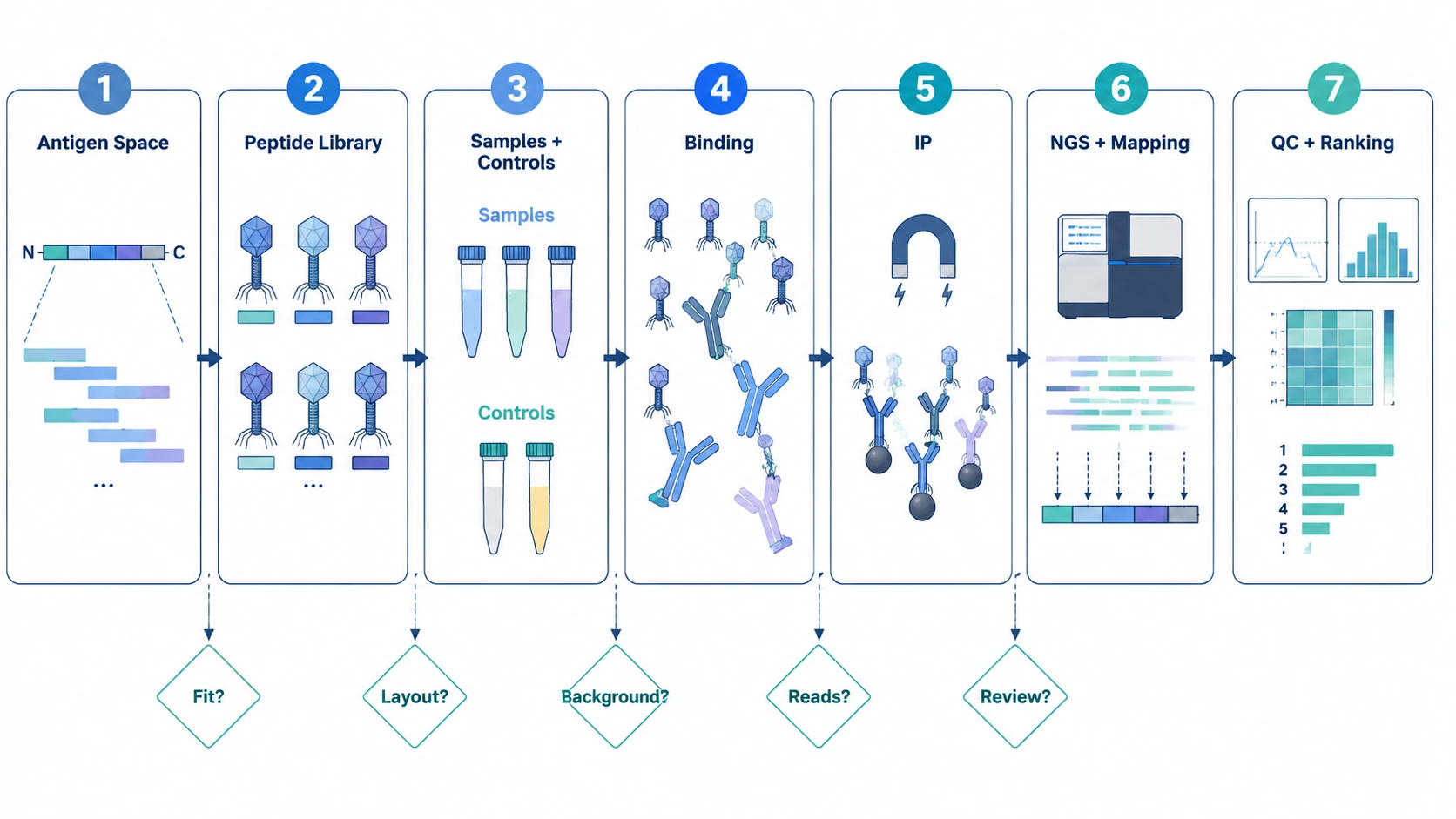
A useful PhIP-Seq workflow diagram is not just a sketch of lab steps. It is a project map. Each stage should answer a practical question. Does the library cover the relevant biology? Are serum or plasma samples compatible with the assay layout? Do controls show nonspecific binding? Are reads mapped cleanly to the input library? Do enrichment scores point to reproducible peptide-level signals, or to batch artifacts and background?
Practical Pain Point: When a PhIP-Seq Study Is Hard to Interpret
A common case starts with organized serum or plasma cohorts and a broad antibody profiling question. The team wants to screen many antigens, variants, or proteome regions instead of testing a short target list by ELISA. Phage immunoprecipitation sequencing is attractive because it connects antibody binding to large numbers of displayed peptide sequences.
The risk usually appears during planning. The team may still be deciding between a proteome-wide peptide library, pathogen-focused peptide library, allergen panel, autoantigen set, or custom peptide library. Sample volume, dilution, freeze-thaw history, cohort balance, and replicate plans may not be locked. Controls sometimes get added late, after the library and batch layout are already fixed.
That is when the final output becomes hard to use. Enriched peptides may cluster in unexpected regions. Negative controls may show background signal. Read counts may vary across samples. Batch effects can start to look like biology. More sequencing cannot repair a poorly matched library or a missing control design. The workflow needs firmer checkpoints before the assay starts.
Root Causes Behind Unclear PhIP-Seq Results
Library and Antigen Space Mismatch
The most common planning problem is a peptide library that does not fit the study question. Peptide length, peptide overlap, antigen tiling density, variant representation, isoform selection, and control peptides all affect which antibody-peptide binding events can be observed. A pathogen-focused peptide library may fit exposure questions. A custom peptide library may be better for engineered proteins, selected antigen families, or defined variant panels.
Weak Control and Cohort Layout
Negative control sample selection, bead-only controls, mock IP planning, randomization, and replicate placement shape the background model. If cases and controls are processed in separate batches, enrichment patterns may track handling order instead of biology. If controls do not match the sample matrix and cohort structure, fold enrichment and Z-score calculations become less stable.
Capture Conditions That Shift Background
During incubation and immunoprecipitation, antibodies bind displayed peptides in the phage-displayed peptide library. Protein A/G beads, or another antibody-capture strategy suited to the species and antibody class, then enrich antibody-bound phage. Incubation time, bead type, washing stringency, and nonspecific binding controls affect how much signal and background remain. Gentle washing can leave nonspecific phage. Overly stringent washing can reduce weaker but reproducible interactions.
Sequencing, Mapping, and Interpretation Limits
Phage DNA amplification and next-generation sequencing convert enriched phage into counts. Barcode demultiplexing quality, index balance, read depth distribution, mapping rate to the peptide library, and read-count reproducibility all affect analysis. Low read counts, uneven barcode performance, or peptide dropout can distort enrichment score estimates.
PhIP-Seq reports linear peptide-level enrichment. It does not directly measure absolute antibody concentration, fully resolve conformational epitopes, or prove clinical relevance on its own. Cross-reactivity can occur when related antigens share short motifs, so candidate antigen prioritization should account for overlapping peptide support, cohort consistency, biological plausibility, and orthogonal validation needs.
Related Services
Main Service
Supporting Service
Validation Service
Alternative Service
Workflow Optimization Guide: A Text-Based PhIP-Seq Workflow Diagram
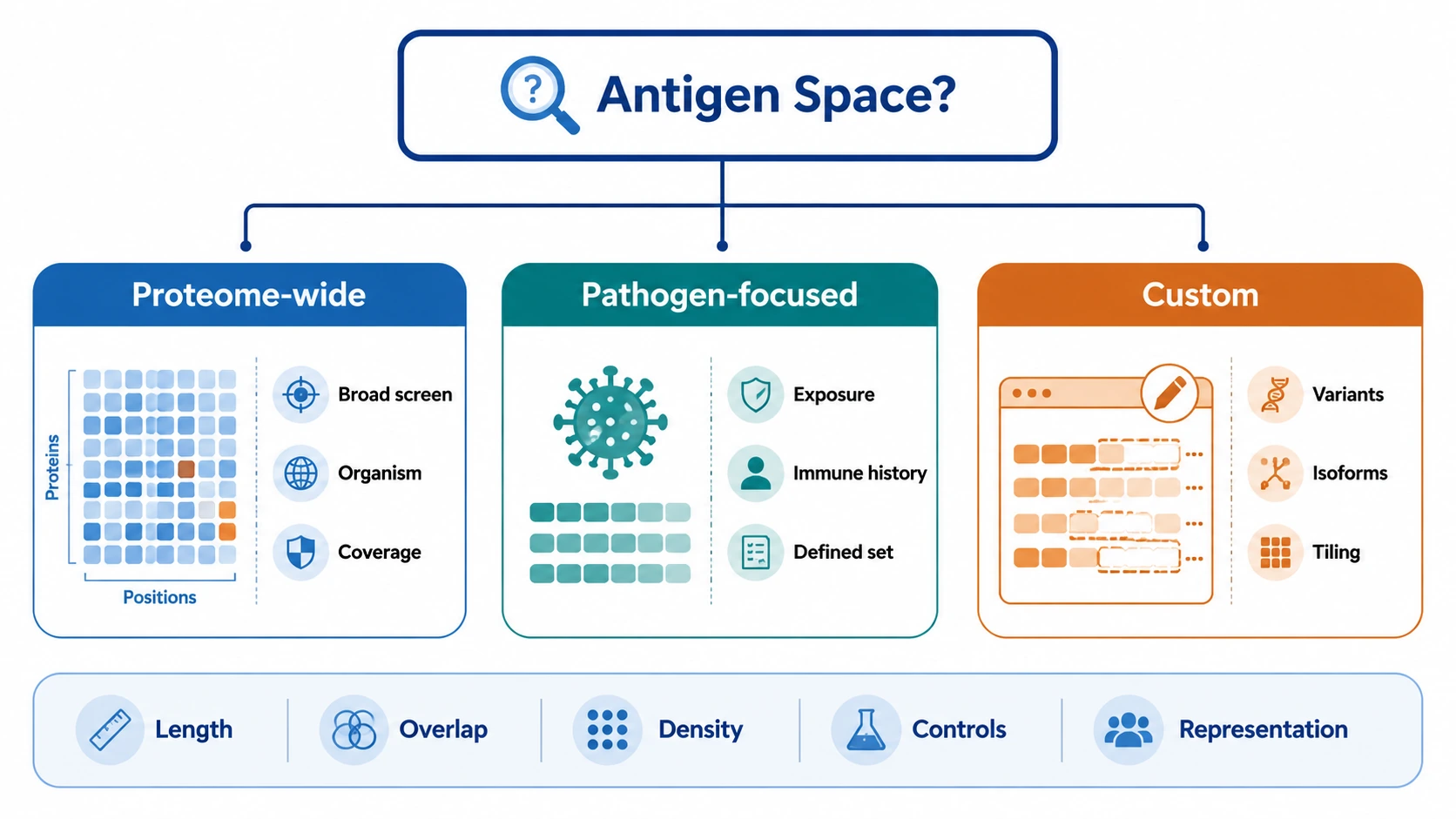
Step 1: Define the Decision the Study Must Support
Start by naming the decision the study should inform. Examples include exposure profiling, autoantigen discovery, vaccine response comparison, allergy-related screening, antibody repertoire comparison, or candidate antigen prioritization. This step is complete when the team can state the intended output: peptide-level ranking, antigen-region mapping, cohort-level comparison, or candidate selection for follow-up.
The bottleneck is often not the sequencing run. It is the mismatch between the study question and the library, control design, or interpretation plan. A clear objective keeps the workflow from becoming a broad screen with no agreed decision criteria.
Step 2: Match Peptide Library Design to the Antigen Space
Choose between a standard library and a custom peptide library by asking what antigen space must be represented. A proteome-wide peptide library fits broad organism-level screening. A pathogen-focused peptide library fits defined infectious exposure or immune history questions. A custom peptide library fits engineered proteins, specific variants, selected isoforms, rare antigen panels, or targeted antigen tiling.
Review peptide length, peptide overlap, tiling density, sequence redundancy, variant coverage, and control peptide inclusion. Check library diversity and library representation using the input library sequencing profile. Low-abundance clones need attention because weak baseline representation can reduce confidence in downstream enrichment.
If your team needs help matching the study objective to a standard or custom library, you can submit your requirements to MtoZ Biolabs for PhIP-Seq workflow evaluation, including antigen selection, peptide library design, control planning, and expected data deliverables.
Step 3: Control Sample Variables and Batch Layout
Before incubation, define sample type, input volume range, dilution strategy, freeze-thaw history, cohort balance, and replicate plan. Serum antibody profiling and plasma antibody profiling can both be considered, but compatibility should be checked against the assay plan and matrix conditions.
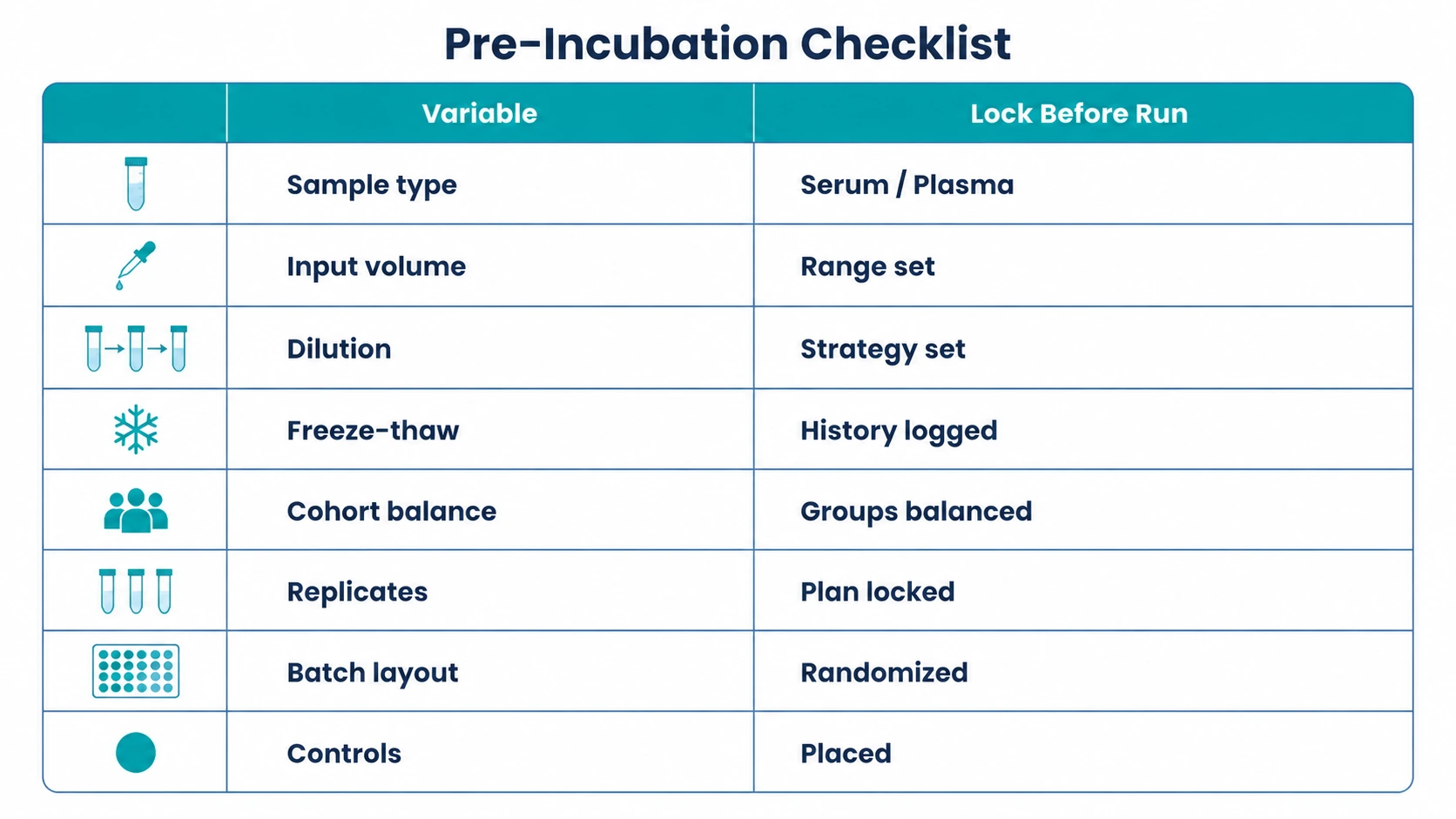
Randomize samples across processing batches when possible. Place negative control samples, technical replicates, and relevant reference samples across the layout instead of grouping them in one batch. This step is complete when sample identifiers, cohort labels, barcodes, replicate positions, and control positions are locked before wet-lab work begins.
Step 4: Tune Antibody-Peptide Binding and Immunoprecipitation Conditions
During incubation, antibodies interact with displayed peptides in the phage-displayed peptide library. The goal is to preserve informative antibody-peptide binding while limiting nonspecific interactions. Incubation time, sample dilution, buffer conditions, and mixing should stay consistent across samples.
Immunoprecipitation then captures antibody-bound phage, commonly using Protein A/G beads when appropriate for the sample species and antibody class. Include bead-only control or mock IP conditions when background binding is a concern. Washing stringency should reduce nonspecific phage without erasing weak but reproducible binding patterns.
Step 5: Establish Sequencing and Mapping QC Before Enrichment Analysis
After washing and recovery, phage DNA amplification and sequencing library preparation convert enriched phage pools into next-generation sequencing readouts. Plan barcode balance and index structure carefully, especially in larger cohorts.
Review barcode demultiplexing, per-sample read count distribution, mapping rate to the designed peptide library, and read-count reproducibility before calculating final enrichment. Compare each sample against the input library baseline so library representation is not mistaken for sample-specific binding. Peptide dropout, strong index imbalance, or low mapping rates should be flagged before biological interpretation starts.
Step 6: Review QC Checkpoints Before Calling Enrichment
A workflow is ready for interpretation when QC supports the analysis model. Check input library representation, negative-control background, bead-only control signal, mock IP behavior, replicate concordance, batch effect patterns, and sample outlier status.
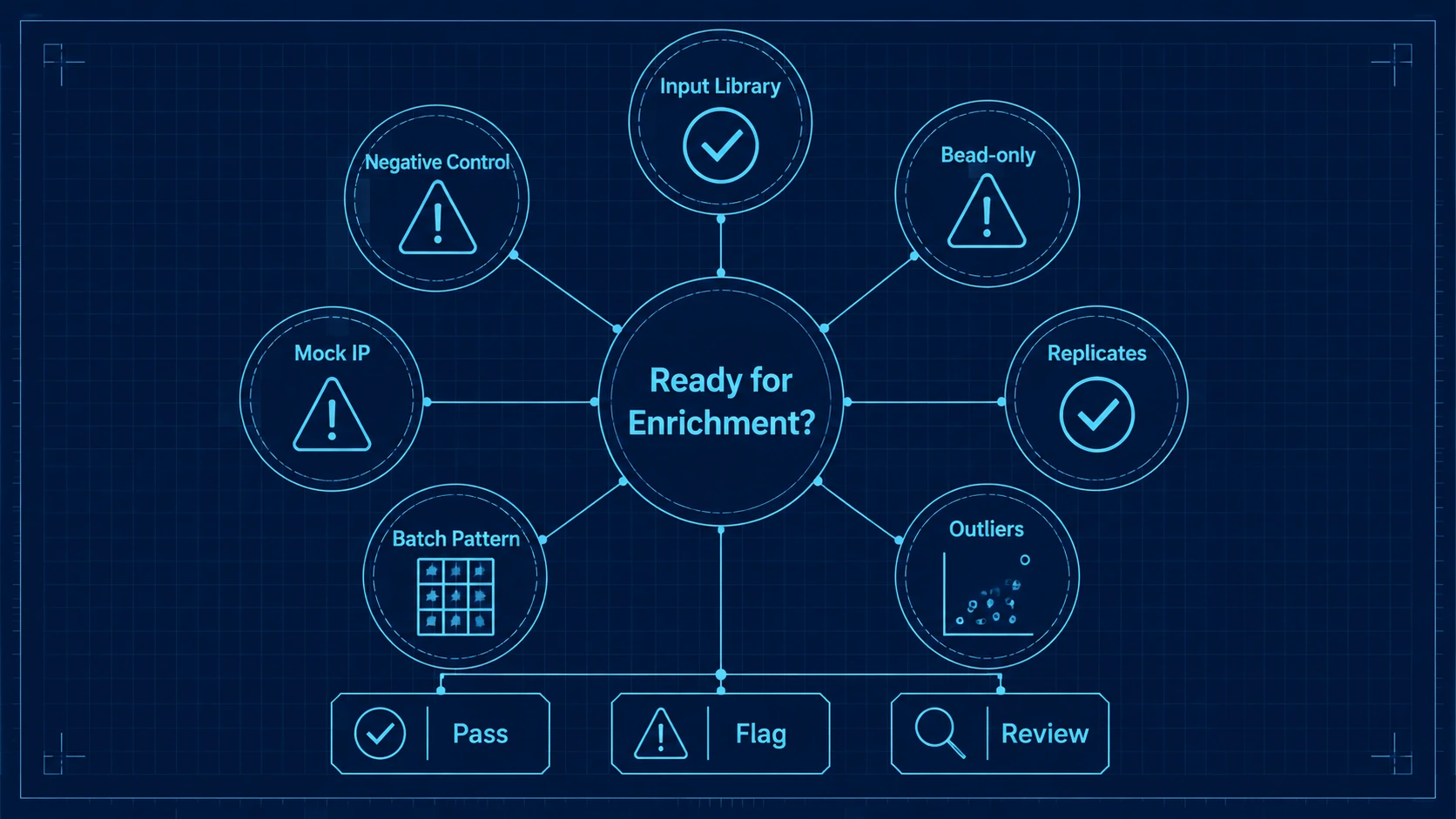
Improvement should show up as cleaner control separation, more consistent read distributions, stronger replicate concordance, and fewer unexplained sample outliers. Enrichment signals should then be assessed by normalized enrichment score, fold enrichment versus controls, Z-score or another statistical enrichment model, false discovery rate strategy, and consistency across related peptides.
Step 7: Interpret Peptide-Level Results and Prioritize Validation
Rank enriched peptides by statistical strength, background behavior, overlap with adjacent peptides, cohort association, and antigen context. Overlapping enriched peptides can support epitope-region mapping, but they should not be treated as complete proof of a conformational epitope. Cross-reactivity review is needed when motifs are shared across related proteins, pathogens, isoforms, or protein families.
For biomarker development, mechanism studies, or translational programs, plan orthogonal validation. ELISA validation may fit targeted antigen follow-up. Peptide array validation can test selected linear peptide regions. Neutralization assay follow-up may be relevant when functional antibody activity is part of the research question. Western blot, targeted immunoassay, or protein array analysis may also help clarify protein-level recognition.
Expected Results and Verification Methods
After workflow optimization, the expected result is not a fixed number of hits. A better sign is that enrichment calls are easier to read against the study question. Negative controls should show a background profile that can be modeled. Input library sequencing should support library representation review. Technical replicates should show replicate concordance suitable for the analysis objective. Read mapping should support peptide-level counting without excessive unmapped reads.
Verification should combine wet-lab and bioinformatics checks. Confirm read count normalization behavior, review fold enrichment and Z-score distributions, evaluate the false discovery rate strategy, and inspect whether related peptides support the same antigen region. Examine outliers before excluding them. If a cohort-level signal depends on one batch, one barcode group, or one extreme sample, treat it as a possible technical pattern until further review supports a biological explanation.
Key Planning Notes Before Running PhIP-Seq
Sample quality and sample amount shape the usable workflow. Limited volume, hemolysis, lipemia, repeated freeze-thaw cycles, or inconsistent storage can affect antibody profiling. If sample supply is scarce, reserve material for validation before using all available volume in discovery screening.
Controls and replicates should be planned as part of the assay, not added after analysis. Negative control sample selection affects the enrichment baseline. Bead-only control or mock IP data help identify nonspecific phage binding. Technical replicates increase sample use and cost, but they can clarify whether borderline enrichment patterns are reproducible.
Batch effect risk rises with large cohorts, multi-day processing, multiple operators, or separated case-control handling. Randomization and balanced batch design reduce avoidable confounding. Contamination control also matters because amplified phage DNA and indexed sequencing libraries can carry over if handling is not disciplined.
Data interpretation has boundaries. PhIP-Seq supports peptide-level interpretation of linear peptide signals. It does not replace calibrated ELISA for absolute antibody concentration, and it does not fully represent conformational epitopes. When the research question requires quantitative titer measurement, folded protein recognition, functional activity, or clinical correlation, follow-up assays should be included in the plan.
Conclusion
A useful PhIP-Seq workflow diagram connects decisions, not only procedures. Interpretable results depend on matching the phage-displayed peptide library to the antigen space, controlling sample and batch design, monitoring immunoprecipitation background, checking sequencing and read mapping quality, and reviewing enrichment with clear peptide-level boundaries.
PhIP-Seq fits discovery-scale antibody profiling when a project needs broad peptide coverage and candidate antigen prioritization. It is less suitable as a standalone answer for absolute antibody quantitation, conformational epitope proof, or clinical diagnostic classification. For studies involving serum or plasma cohorts, custom antigen panels, complex controls, or validation planning, contact MtoZ Biolabs to discuss the PhIP-Seq workflow, sample conditions, sequencing QC, enrichment analysis, and follow-up assay options for your project.
FAQ
What should I prepare before requesting a PhIP-Seq project review?
Prepare the study objective, species, sample type, cohort groups, approximate sample number, available sample volume, antigen list or target organism, preferred library type, control samples, and expected deliverables.
Can PhIP-Seq use archived serum or plasma samples?
Archived samples may be usable if storage history, freeze-thaw exposure, volume, and matrix quality are acceptable for the planned workflow. Samples with inconsistent handling should be flagged before layout design.
Is a custom peptide library always better than a standard library?
No. A standard library can be more efficient when it already covers the antigen space. A custom peptide library is more appropriate when variants, engineered sequences, selected isoforms, or a focused antigen panel drive the study question.
How do I know whether an enriched peptide is biologically relevant?
Look for reproducibility, low control background, support from overlapping peptides, consistency within the cohort, plausible antigen context, and reduced likelihood of cross-reactivity. Strong candidates still need orthogonal validation.
When should I choose ELISA validation after PhIP-Seq?
Use ELISA validation when you need targeted confirmation against a defined antigen or peptide and want a focused assay for selected samples. PhIP-Seq can nominate candidates; ELISA can test a narrower hypothesis.
Can deeper sequencing fix weak PhIP-Seq results?
Deeper sequencing may help when read depth is the limiting issue. It cannot correct a poorly matched peptide library, weak sample design, high nonspecific binding, missing controls, or unresolved batch effects.
How to order?

