





Peptide De Novo Sequencing: When Database Search Is Not Enough in LC-MS/MS Identification
- the precursor ion is reasonably clean,
- fragment ions are rich enough to support a sequence tag,
- b ions or y ions show at least partial continuity,
- routine database search settings have already been reviewed,
- and the remaining explanation points to sequence novelty, a sequence variant, truncation, non-canonical peptide structure, or PTM complexity.
- spectral quality is poor,
- co-fragmentation is obvious,
- precursor isolation was broad,
- the spectrum is sparse or noisy,
- or the original database search was too narrow to test the expected biology.
- a sequence tag,
- one or more candidate peptide backbones,
- annotated fragment-ion evidence,
- an explanation of where confidence is strong or weak,
- and a shortlist of likely sequence variant, truncation, or PTM scenarios.
- targeted LC-MS/MS on the same precursor ion,
- repeat acquisition with improved isolation,
- alternate fragmentation where appropriate,
- synthetic peptide comparison,
- MRM or PRM confirmation,
- or orthogonal validation tied to the project goal.
SEO Title: Peptide De Novo Sequencing: When Database Search Is Not Enough in LC-MS/MS Identification
Primary Keyword: peptide de novo sequencing
Article Template: informational-science-article
When an LC-MS/MS dataset contains a real, interpretable peptide signal but database search still returns a no-hit spectrum, a weak peptide-spectrum match (PSM), or conflicting annotations, peptide de novo sequencing is often the right next step. Search failure alone is not the deciding factor. What matters is the gap between solid tandem mass spectrometry evidence and a reference space that no longer explains it.
Quick Decision Guide
Escalate to peptide de novo sequencing when:
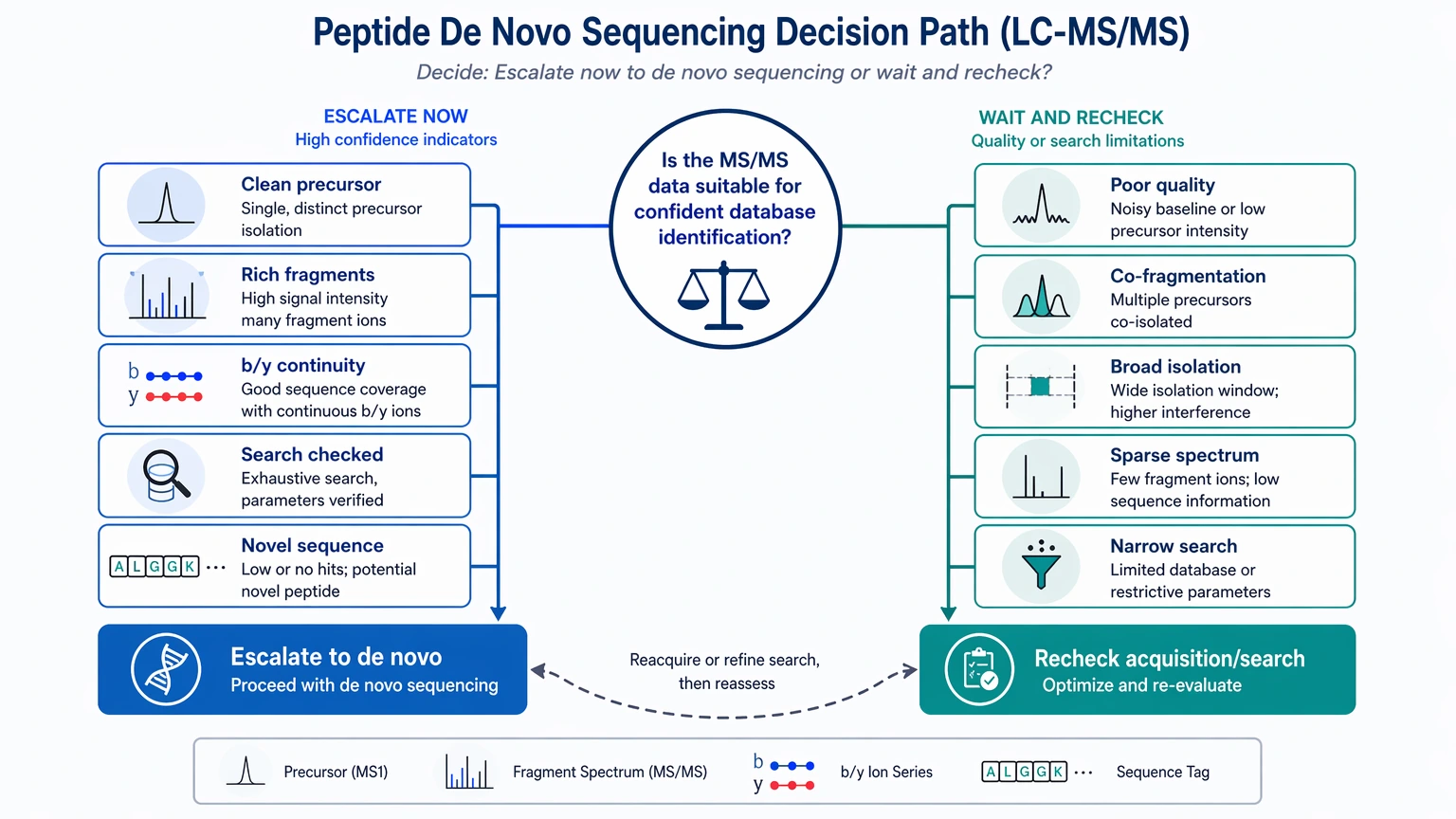
Do not escalate yet when:
What Peptide De Novo Sequencing Means in LC-MS/MS
Peptide de novo sequencing infers amino acid order directly from LC-MS/MS fragment ions instead of relying only on a database search against known sequences. In tandem mass spectrometry, a selected precursor ion is fragmented, and the resulting ions are measured by mass-to-charge ratio (m/z). De novo interpretation then uses mass differences between fragment ions, especially b ions and y ions, to reconstruct part or all of the peptide backbone.
That is the key difference from database search. A database search compares an observed MS/MS spectrum with theoretical spectra generated from a defined sequence space. It works well when the true peptide is already present in the searched FASTA, when enzyme assumptions fit the sample, and when expected post-translational modification (PTM) settings are included. It becomes much less dependable when the peptide sequence is absent, altered, heavily modified, or processed in an unexpected way.
So peptide de novo sequencing is not a universal replacement for database search. It is an escalation strategy for unknown peptide identification when the database has become the limiting part of the workflow.
Why a Real Peptide Can Still Fail Database Search
A failed database search does not automatically mean the peptide is novel. In practice, four evidence patterns are more informative than a simple hit-versus-no-hit view.
First, the searched reference may not contain the true sequence. This comes up often in non-model organisms, engineered constructs, natural peptide discovery, venom studies, and impurity work where an unexpected species is present but missing from the database.
Second, the peptide backbone may break routine search assumptions. Truncation, non-tryptic processing, sequence variant formation, cyclization, and other non-canonical peptide features can leave a genuine spectrum hard to map under standard digestion logic.
Third, PTM burden can disrupt otherwise reasonable search workflows. Multiple PTMs, uncommon PTMs, or uncertain PTM localization expand the search space and weaken scoring. In that situation, the issue is not only modification assignment. It is also backbone recognition.
Fourth, the search output can look worse than the spectrum itself. When a spectrum shows coherent fragment ions and usable mass gaps, yet the best database hits remain weak or biologically implausible, the reference-dependent method is probably the bottleneck.
Signs That the Problem Is the Search Space, Not the Spectrum
The main decision is whether you are dealing with a database limitation or a data-quality limitation. The table below separates those cases.
| Scenario | Recommended workflow | Main interpretation risk | Best next step |
|---|---|---|---|
| Clean precursor ion, rich fragment ions, no-hit spectrum | Extract a sequence tag and begin peptide de novo sequencing | Full sequence confidence may still be incomplete | Use targeted follow-up if the sequence will drive a decision |
| Low-confidence PSM with several inconsistent top hits | Recheck search parameters, then compare with de novo interpretation | Search ambiguity may reflect mixed candidate explanations | Reacquire if isolation quality was marginal |
| Suspected sequence variant or truncation product | Combine database search with peptide de novo sequencing | Protein-level mapping may remain partial | Validate variant-specific fragment evidence |
| PTM-rich peptide with unexplained precursor mass | Separate backbone inference from PTM assignment | PTM localization may remain uncertain | Add orthogonal validation for critical sites |
| Sparse or chimeric spectrum | Improve acquisition before de novo work | De novo interpretation will inherit the same ambiguity | Repeat LC-MS/MS with cleaner isolation |
The practical takeaway is straightforward: escalate when the spectrum is informative and the search space is not.
Service Routes to Consider
For this project scenario, readers usually compare these service routes before requesting a quote or submitting samples.
What Spectral Evidence Supports Credible De Novo Interpretation
Not every MS/MS spectrum can support peptide de novo sequencing. The most useful spectra usually share a few recognizable features.
A reasonably isolated precursor ion lowers the risk of co-fragmentation and reduces the chance that the spectrum is chimeric. Continuous b ions or y ions across several residues support ordered sequence inference. Coherent mass gaps that match amino acid composition help build a sequence tag. Internal fragment support can strengthen difficult regions, although it should not be the only evidence. Good sequence coverage at one terminus may still be enough to propose a candidate backbone even when the opposite terminus remains incomplete.
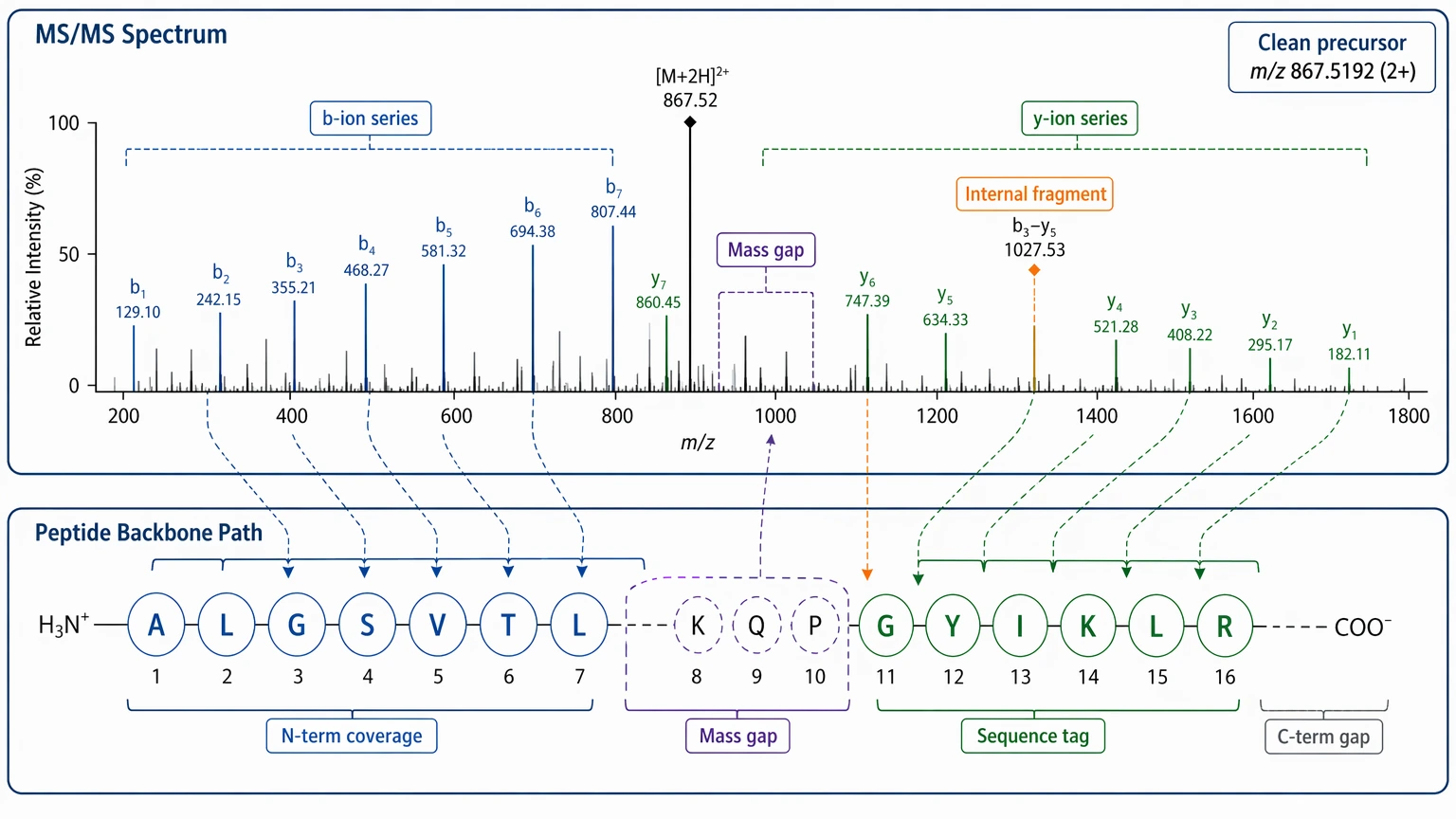
Even with a strong spectrum, some uncertainty can stay on the table. Leucine/isoleucine ambiguity often remains in standard LC-MS/MS workflows because those residues are isobaric. PTMs add another layer of uncertainty when modification identity, multiplicity, or PTM localization is not fully resolved. Put simply, peptide de novo sequencing can yield a well-supported sequence proposal without guaranteeing residue-by-residue certainty at every position.
A useful checkpoint is whether the spectrum can support a defensible sequence tag first. If it cannot, a full de novo interpretation is unlikely to become reliable just because the algorithm changed.
When Peptide De Novo Sequencing Is Most Justified
Peptide de novo sequencing is most useful in a fairly specific set of project types.
It is often justified when the sample may contain a sequence variant missing from the reference FASTA. It is also a strong option when truncation, unexpected cleavage, or non-enzymatic processing is suspected. PTM-rich peptides can justify escalation when routine variable modification settings no longer account for the precursor mass. The same applies to non-canonical peptides from poorly annotated organisms or discovery programs where reference databases are incomplete by design.
By contrast, if the main issue is weak spectral quality, de novo work should usually wait until the acquisition problem is fixed. Good interpretation starts with interpretable fragmentation, not with a more aggressive search strategy.
If your team has reached that point and needs to decide whether the current dataset supports sequence reconstruction, you can submit your requirements or evaluate your project with MtoZ Biolabs using the raw files, search results, and sample context instead of relying on another round of trial-and-error reprocessing.
Expected Results and Validation Methods
A de novo project does not always start with a final sequence call. The first deliverables are usually analytical rather than absolute.
Immediate deliverables may include:
These outputs help determine whether the unknown feature is truly novel, altered, or simply mismatched to the database search space.
Follow-up confirmation may include:
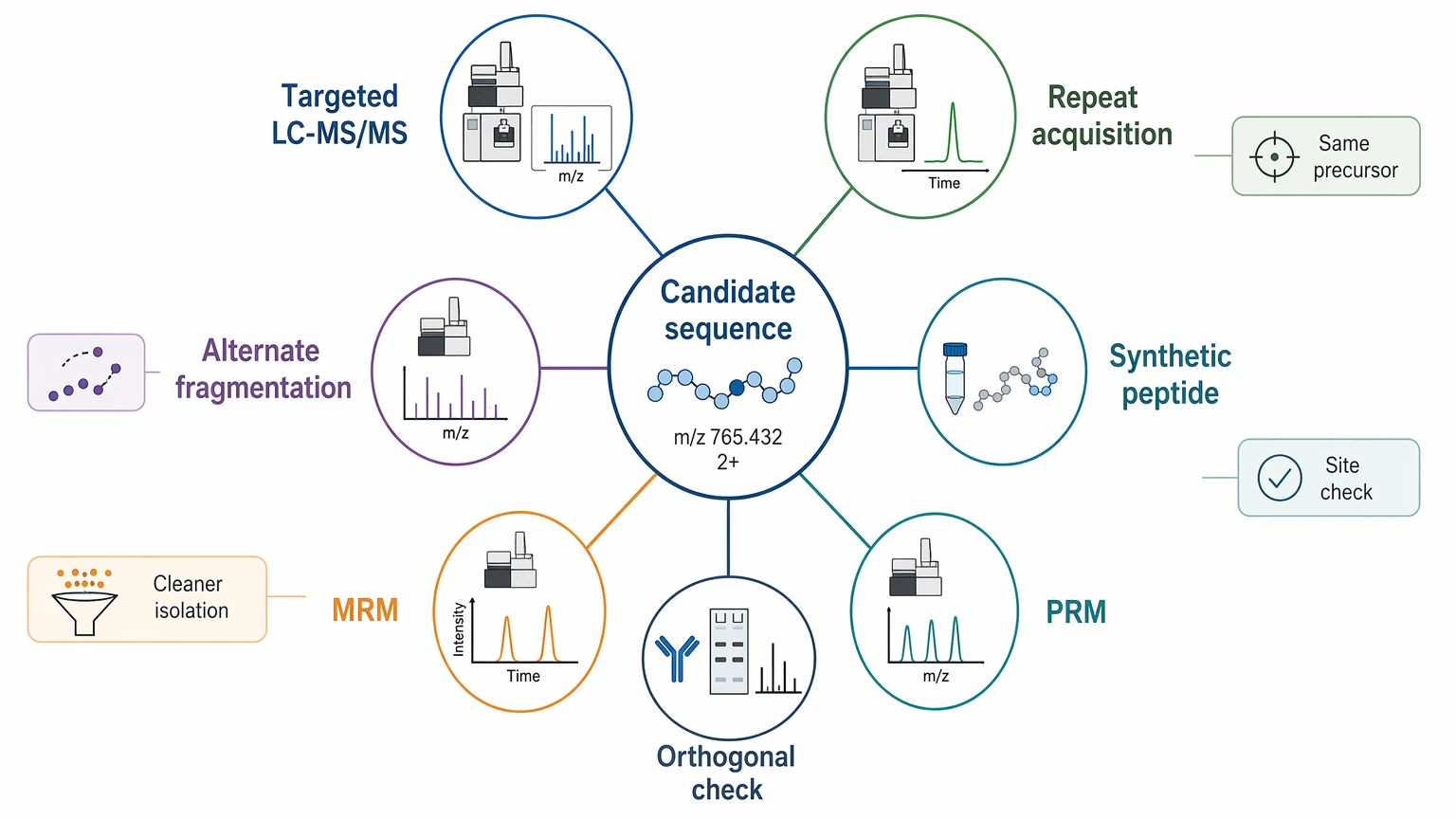
That distinction matters. A candidate sequence proposal is an analytical result. Confirmation still needs another step when the answer will support a publication claim, impurity assignment, construct verification, or another high-consequence decision.
Key Cautions and Practical Limits
Peptide de novo sequencing operates within clear analytical boundaries.
Sample quality and sample amount matter because repeat acquisition is often necessary when fragment-ion coverage is incomplete. If only trace material remains, confirmation options can narrow fast.
Controls and repeat expectations matter as well. A single unexplained spectrum can be informative, but repeated detection of the same precursor ion increases confidence that the feature is real rather than an acquisition artifact.
Batch effects and contamination risk also deserve attention. Carryover, mixed isolation windows, and background peptides can create misleading fragment patterns, especially in low-abundance workups.
Interpretation boundaries need to stay explicit. Peptide de novo sequencing can support a strong sequence hypothesis, but LC-MS/MS alone may not resolve every ambiguity. Leucine/isoleucine ambiguity, PTM localization uncertainty, and incomplete terminal coverage can remain. Database-search limits also continue to matter after de novo work starts, because homolog mapping and downstream annotation still depend on available reference information.
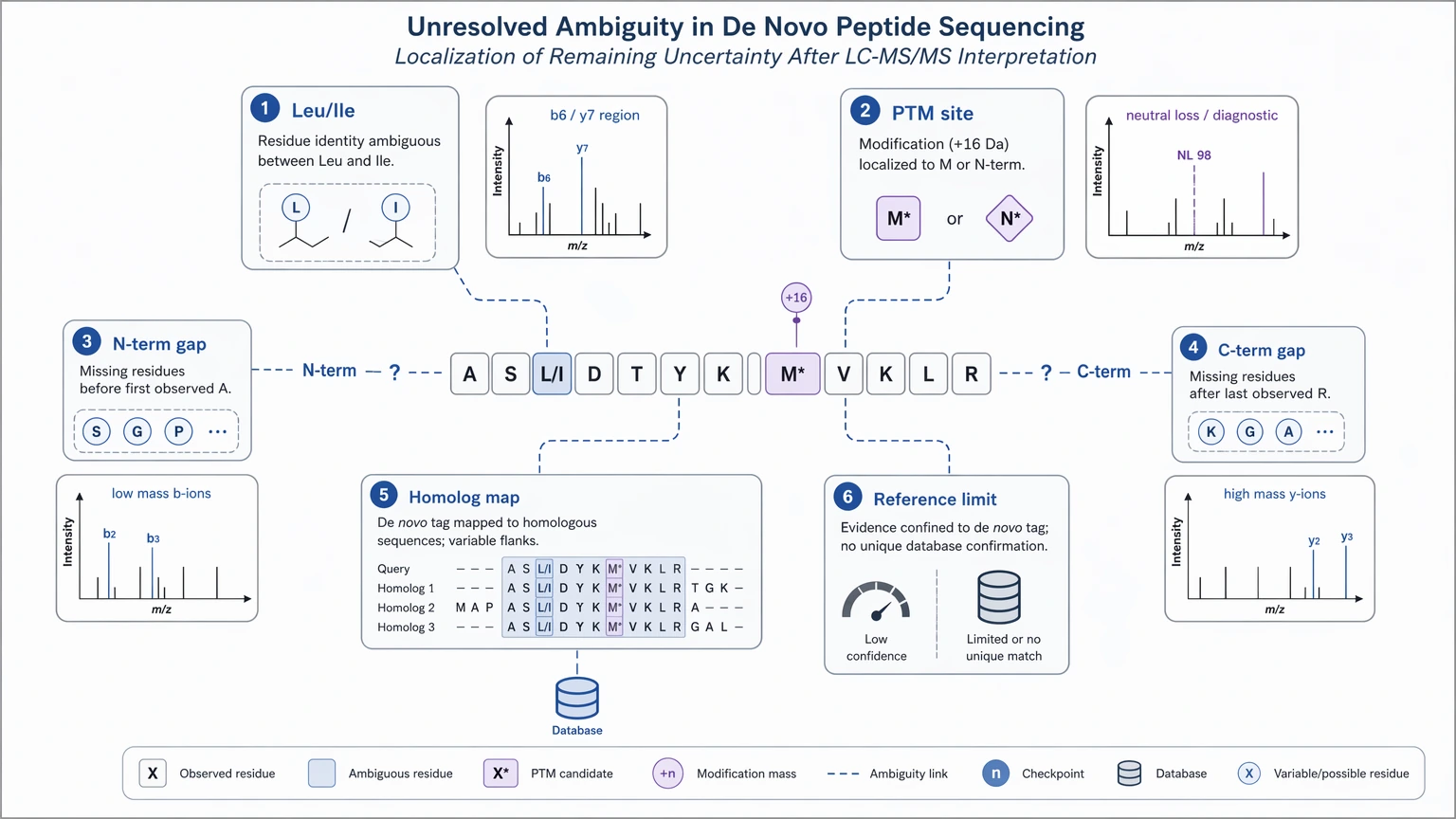
Another method may be the better next step when the real issue is poor precursor isolation, a highly chimeric spectrum, a complex mixture with insufficient separation, or a question that depends more on targeted confirmation than backbone discovery. In those cases, repeat LC-MS/MS, targeted validation, or a different analytical strategy may be more efficient than forcing a low-confidence de novo call.
What to Prepare Before Requesting Evaluation
Teams usually get a faster, more useful assessment when they provide a compact data package. Helpful inputs include the raw LC-MS/MS files, the precursor ion m/z and charge state, retention time, the original database search settings, the unsatisfactory PSM output, and a short note explaining why the current identification is not credible.
It also helps to state the project goal plainly: Do you need a sequence tag, a candidate full sequence, a sequence variant assessment, PTM-aware interpretation, or validation planning? If you want a technical review of workflow fit before using more sample, contact MtoZ Biolabs with the available data and sample constraints so the team can evaluate the project and recommend the most suitable confirmation path.
Conclusion
Peptide de novo sequencing becomes the right escalation point when LC-MS/MS has already produced an interpretable spectrum, but database search cannot explain that spectrum with a credible peptide-spectrum match. This pattern shows up most often in sequence variant discovery, truncation analysis, PTM-rich peptides, non-canonical peptide work, and samples that fall outside well-covered reference databases. In that setting, the goal is not to replace database search across the board, but to move from reference matching to evidence-based sequence inference, then confirm the result with the level of follow-up the project actually requires. For unknown peptide findings that need sequence reconstruction, project-fit review, or validation planning, contact MtoZ Biolabs to discuss the raw data, sample limits, and decision target before committing to the next experiment.
FAQ
Can peptide de novo sequencing be useful if I only need a partial answer?
Yes. In some projects, a sequence tag is enough to distinguish a truncation product, support a suspected sequence variant, or narrow the candidate space before targeted confirmation.
Does fragmentation mode affect whether de novo work is realistic?
Yes. CID, HCD, and ETD can produce different fragment-ion patterns, and some peptides become easier to interpret with one fragmentation strategy than another. The best choice depends on peptide chemistry and the type of ambiguity you need to resolve.
How does a chimeric spectrum affect de novo sequencing?
A chimeric spectrum can mix fragment ions from more than one precursor ion. That can create false residue paths and lower confidence in any proposed sequence, even when one component peptide is real.
Should I rerun the database search before escalating?
Usually yes, but only as a focused check. Review enzyme assumptions, PTM settings, precursor tolerances, and reference coverage first. If those settings were already reasonable and the spectrum still lacks a credible match, escalation is justified.
Is peptide de novo sequencing the right choice for every unknown MS/MS feature?
No. It is best suited to cases where the spectrum itself supports sequence inference. If the main problem is weak signal, contamination, or poor isolation, acquisition improvement or targeted follow-up is often the better next step.
How to order?

