





iTRAQ LC-MS/MS Quantitative Proteomics Workflow: Multiplexing, Accuracy, and Method Selection
- iTRAQ labels peptides after digestion, allowing multiple samples to be mixed and analyzed together in one LC-MS/MS workflow.
- Reporter ions generated in MS/MS provide relative abundance measurements across multiplexed samples.
- Pooling labeled peptides reduces inter-run technical variation compared with separately injected workflows.
- Fractionation, labeling efficiency control, and interference management strongly influence quantitative depth and accuracy.
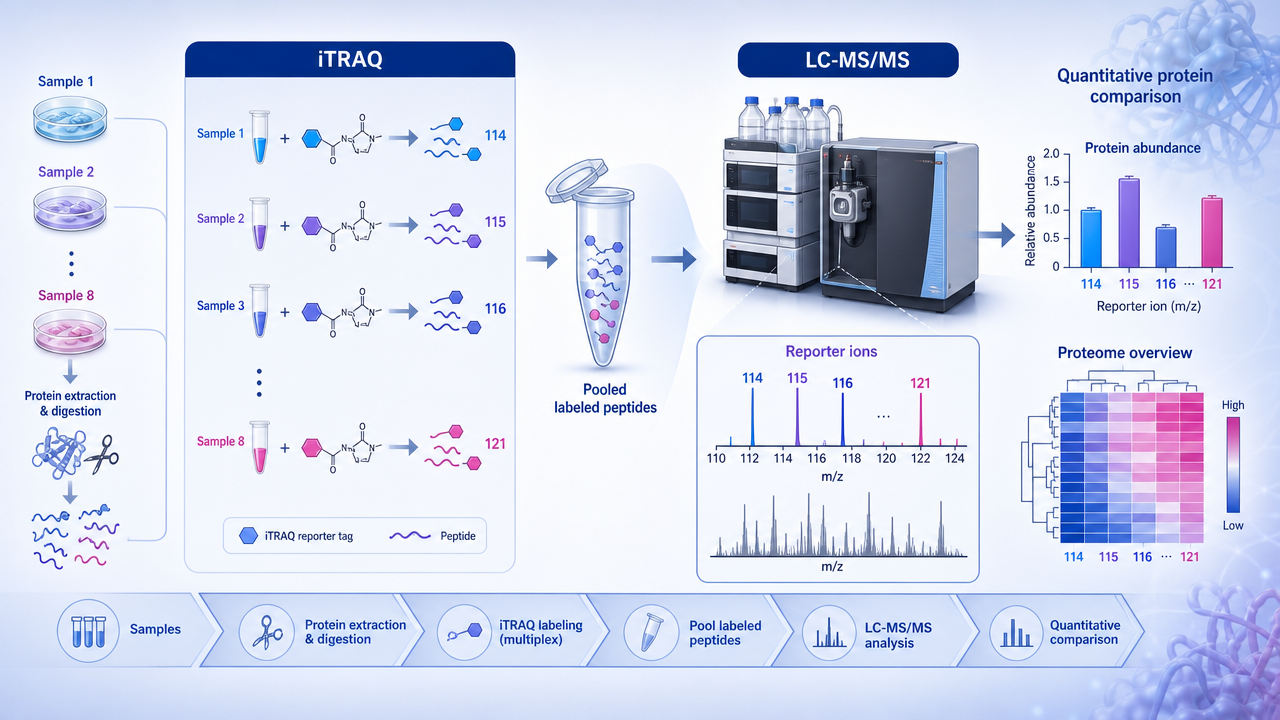
iTRAQ LC-MS/MS is an isobaric labeling workflow for multiplex quantitative proteomics in which peptides from different samples are chemically tagged, pooled, fractionated, and quantified through reporter ions released during MS/MS. The method is especially useful when researchers need consistent relative quantification across several biological groups, including tissues, body fluids, animal models, or treatment-response studies where metabolic labeling is impractical.
Key Takeaways
How iTRAQ Works?
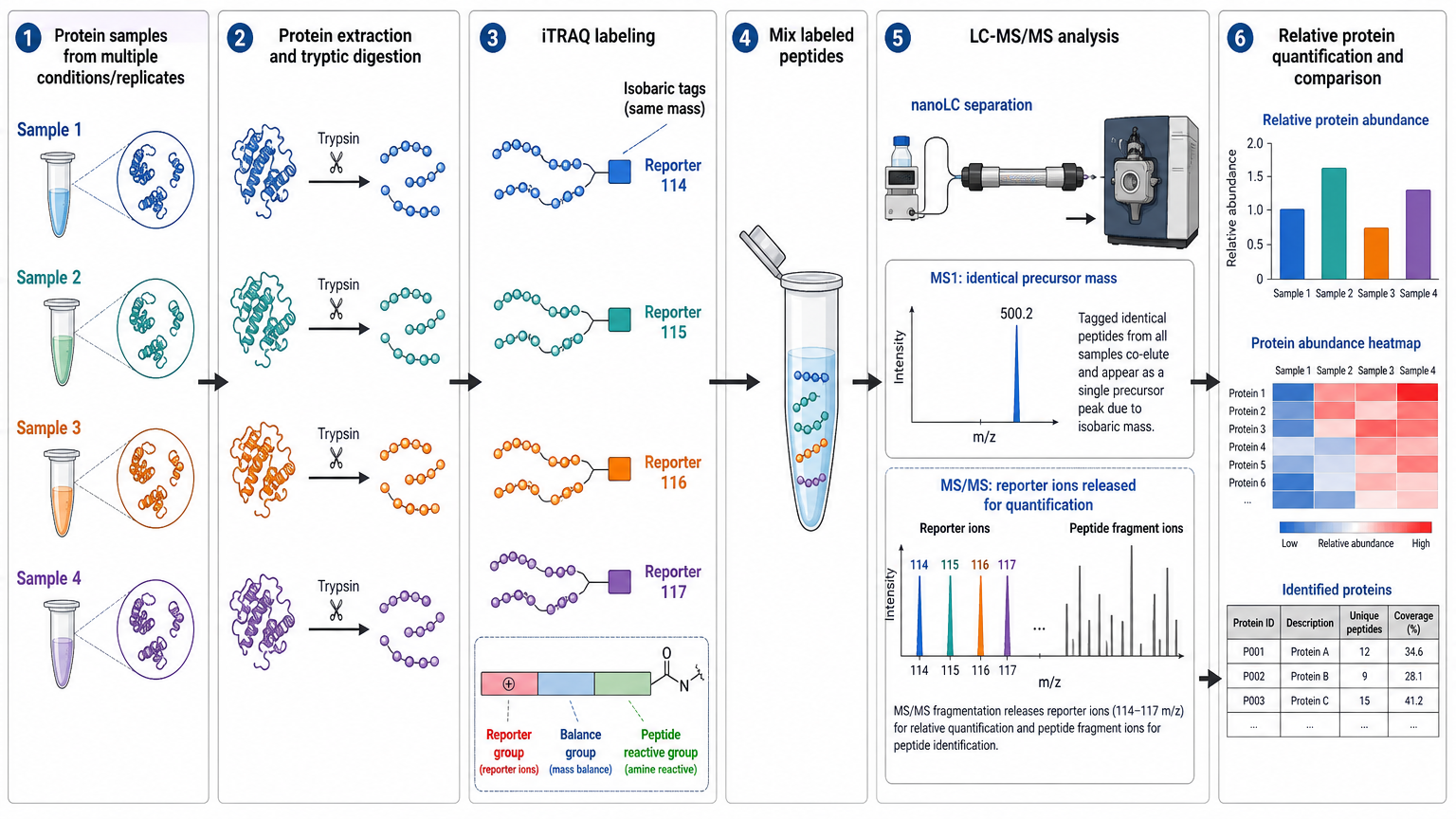
iTRAQ tags are designed so labeled peptides have the same total mass at the MS1 level but produce distinct low-mass reporter ions after fragmentation. Each tag contains a reporter group, a balance group, and a peptide-reactive group that binds peptide N-termini and lysine residues. After labeling, the samples are combined into one mixture and analyzed together.
Related Services
iTRAQ-based Quantitative Proteomics Analysis
iTRAQ/TMT/MultiNotch Quantitative Proteomics Service
TMT/iTRAQ Labeling-Based Quantitative Service
Standard iTRAQ LC-MS/MS Workflow
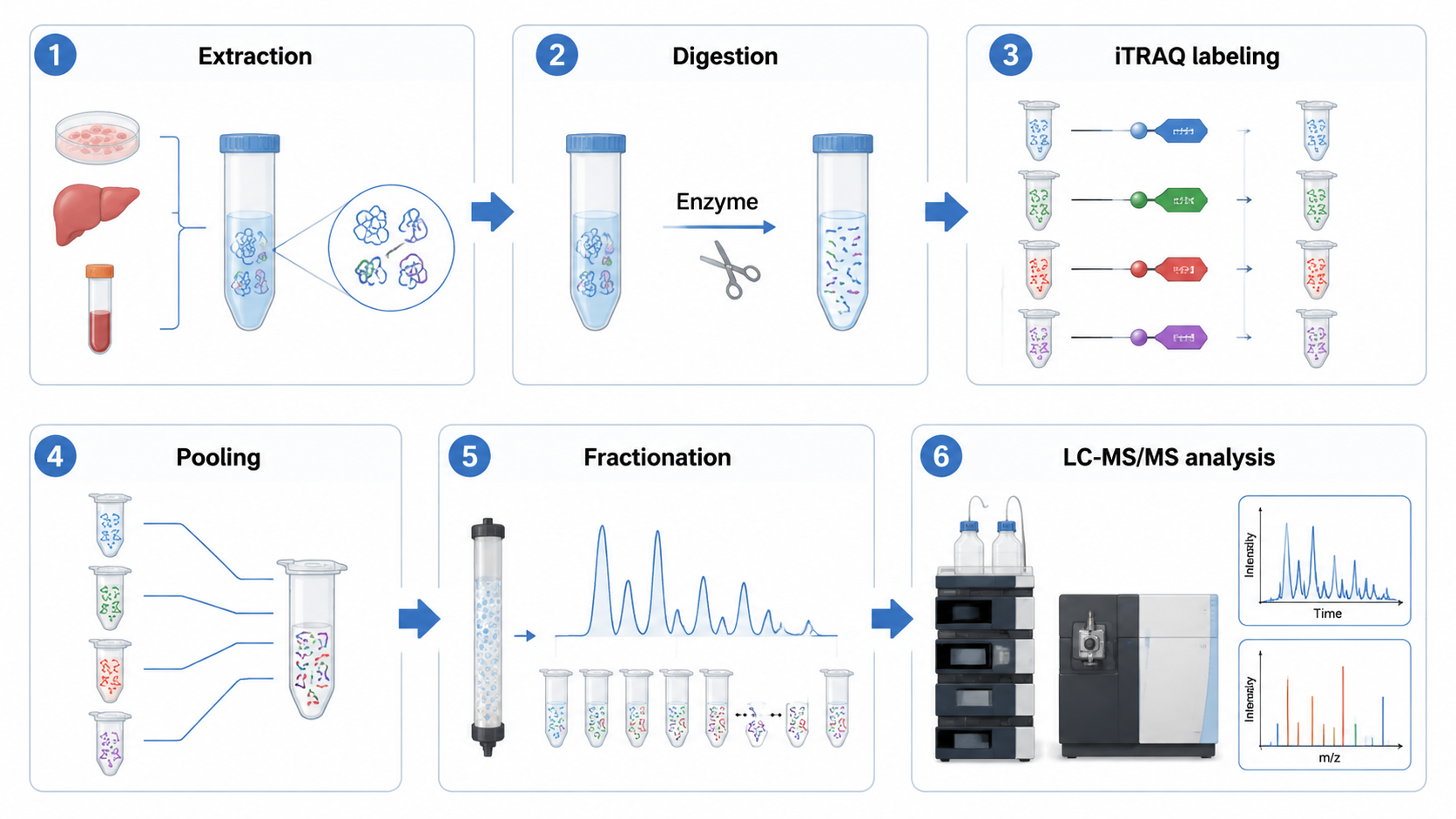
The practical workflow usually includes protein extraction, reduction and alkylation, proteolytic digestion, peptide cleanup, iTRAQ labeling, sample pooling, high-pH fractionation, nanoLC-MS/MS, database searching, reporter ion quantification, normalization, differential expression analysis, and pathway interpretation.
Labeling efficiency must be checked before pooling because incomplete labeling can distort quantitative comparisons. High-pH reversed-phase fractionation is often used to increase proteome depth before LC-MS/MS.
Why Researchers Choose iTRAQ?
1. Multiplexing and Throughput
iTRAQ supports simultaneous comparison of multiple samples in one experiment, which is helpful for time-course designs, drug-response groups, or paired disease-control studies.
2. Reduced Technical Variation
Because labeled samples are pooled before LC-MS/MS, much of the injection-to-injection variability seen in separate runs is reduced.
3. Better Detectability for Low-Abundance Peptides
Pooling can improve apparent MS1 signal strength for peptides that would otherwise be weak in separate single-sample injections, although downstream interference still needs control.
Limitations and Interpretation Risks
| Issue | Why it happens | Practical response |
|---|---|---|
| Ratio compression | Co-isolated precursors contaminate reporter ions | Use narrower isolation and cleaner fractionation |
| Incomplete labeling | Peptides fail to react uniformly | Validate labeling efficiency before pooling |
| Batch comparison limits | Different plexes still require normalization | Use references and balanced design |
| Workflow complexity | Tagging and fractionation add handling steps | Reserve iTRAQ for studies that benefit from multiplexing |
iTRAQ vs Other Quantitative Strategies
iTRAQ is often compared with TMT, SILAC, and label-free proteomics. TMT now provides broader multiplex capacity in many platforms, while SILAC is limited mainly to cell culture systems but can produce very clean quantitative designs. Label-free workflows avoid labeling cost and chemistry, but they depend more heavily on run-to-run reproducibility.

Common Applications
iTRAQ LC-MS/MS is widely used for disease mechanism studies, biomarker discovery, drug response profiling, tissue comparison, developmental biology, and pathway-level differential expression analysis.
FAQ
1. What does iTRAQ measure in proteomics?
iTRAQ measures relative peptide and protein abundance across multiple labeled samples by comparing reporter ion intensities released in tandem mass spectra.
2. Is iTRAQ still useful if TMT exists?
Yes. TMT has expanded multiplexing in many workflows, but iTRAQ remains a valid and well-established option for multiplex quantitative proteomics.
3. What causes ratio compression in iTRAQ?
Ratio compression happens when unrelated co-isolated peptides contribute reporter ion signal, making true abundance differences appear smaller than they are.
Conclusion
iTRAQ LC-MS/MS remains a robust multiplex quantitative proteomics strategy when the study requires pooled analysis, consistent relative quantification, and compatibility with non-culture biological samples. The strongest results come from careful labeling control, clean fractionation, interference-aware MS analysis, and statistics matched to multiplex experimental design.
How to order?

