





How to Perform De Novo Sequencing: Key Steps for Reliable Peptide Identification
-
Is the target purified, gel-enriched, or present in a complex mixture?
-
Is database-assisted confirmation acceptable, or is reference-free analysis required?
-
Are modifications, truncations, or sequence variants expected?
-
Will the result support cloning, publication, comparability testing, or release QC?
-
high mass accuracy on precursor and fragment ions
-
enough scans across chromatographic peaks
-
collision energy suited to the peptide class being analyzed
-
replicate runs when sample amount allows
-
raw file retention for manual re-inspection
Introduction
Researchers often need protein sequence evidence when a construct file, transcript, or database entry is incomplete. A purified band may require confirmation before cloning. A recombinant batch may need QC documentation. An antibody product may lack full genetic records. A protein from a non-model organism may have no reliable reference proteome. In each case, the practical question is the same: how should the sample move from preparation to sequence analysis so the final protein sequencing report can be trusted?
The process is not one fixed protocol. Terminal methods, database-assisted LC-MS/MS, de novo peptide interpretation, and protein-level assembly each fit different reporting goals. Weak results usually trace back to decisions made before data analysis: unclear project scope, poor sample purity, mismatched digestion design, low-quality MS/MS spectra, or software output accepted without review. A dependable workflow treats sequence assignment as evidence building across preparation, acquisition, and analysis stages.
Related Services
| Research Need | Recommended Service Direction |
|---|---|
| MS-based protein sequence confirmation | |
| Full-length sequence recovery | |
| Sequence without reliable database match | De Novo Protein Sequencing Service |
| N-terminal or C-terminal confirmation | N-Terminal Sequencing Service / C-Terminal Sequencing Service |
| Unknown or poorly annotated proteins | Unknown Proteins Sequencing Service |
For projects where sample type, coverage depth, or reporting format is uncertain, MtoZ Biolabs can help match MS-based protein sequencing, terminal sequencing, de novo assembly, or a combined workflow to the biological decision behind the project.
Why Sequencing Projects Fail
Most failed projects share a small set of root causes. The sample may be too complex for the chosen method. The target protein may be present at low abundance beside dominant contaminants. Digestion may be incomplete, producing long peptides that fragment poorly. MS/MS spectra may lack enough consecutive fragment ions to support confident assignment. A database-assisted workflow may be applied when the correct reference sequence is missing or wrong.
Another common issue is treating software output as final proof. Automated peptide-spectrum matching and de novo tools are valuable, but they propose candidates rather than guaranteed truth. Without manual spectrum review, overlap checks, and clear ambiguity labeling, a reported sequence may look complete while leaving critical residues unsupported.
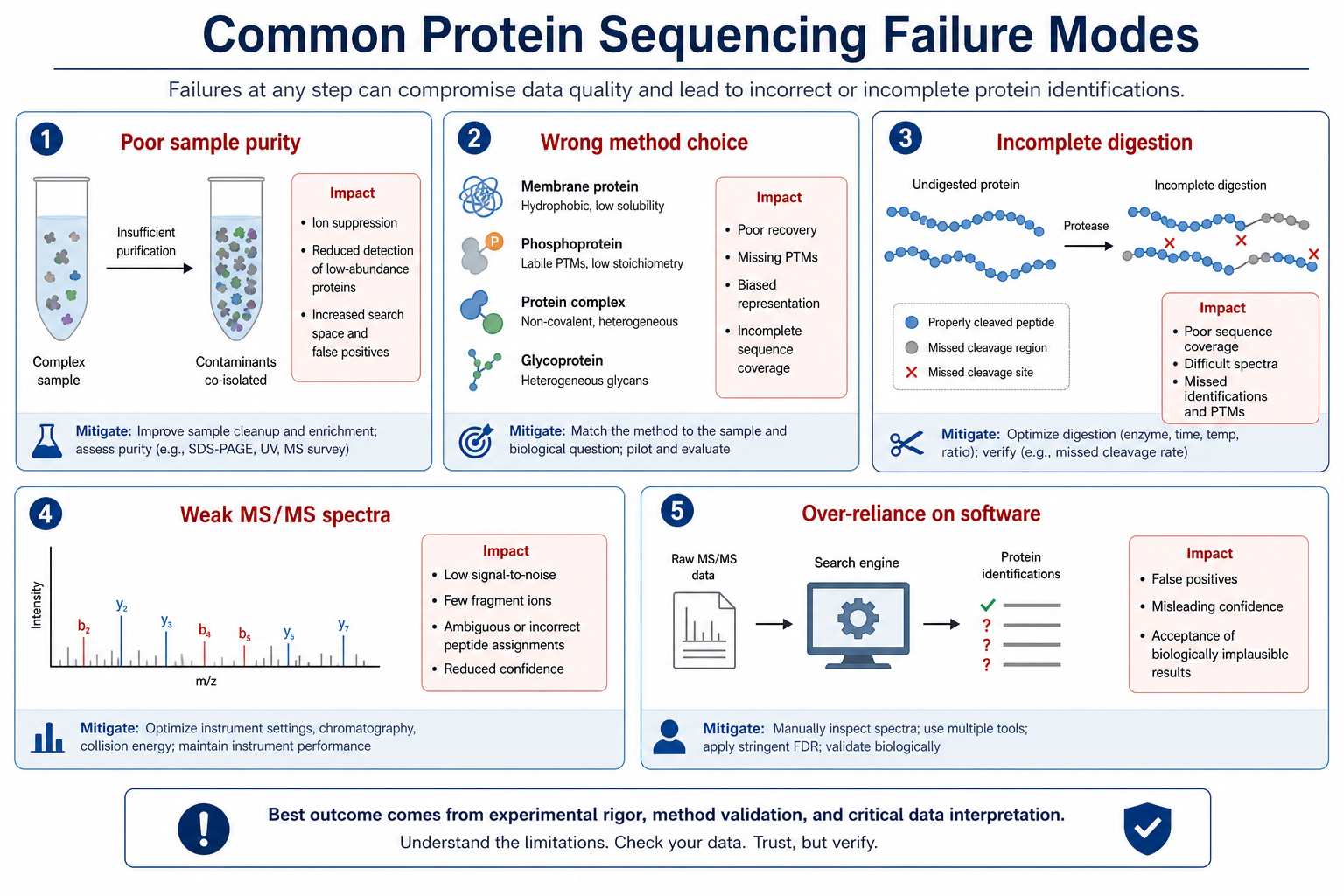
Figure 1. Common reasons sequencing projects produce weak or unreliable sequence evidence
Method mismatch is an overlooked failure mode. Terminal sequencing is efficient for N- or C- terminal confirmation but is not a substitute for full-length coverage. Database search works when a correct reference exists. De novo interpretation is needed when the sample may differ from any available entry. Choosing the wrong path early wastes sample, MS time, and interpretation effort.
Step 1: Define the Sequencing Goal Before Sample Prep
Before digestion or instrument time is committed, define what the project must prove. Some studies need terminal confirmation only. Others need regional peptide coverage. Others require protein-level assembly across a domain, chain, or full-length product.
Useful planning questions include:
A narrow goal improves efficiency. A broad full-length goal without added fractionation, replicate depth, or complementary enzymes often ends in partial coverage and disputed sequence calls.
Step 2: Prepare a Sequence-Friendly Sample
Sample preparation sets the ceiling for downstream sequence analysis. Cleaner input generally produces sharper spectra, fewer ambiguous assignments, and more efficient use of LC-MS/MS acquisition time.
For gel-based samples, excise the band tightly and minimize keratin exposure. For recombinant proteins, confirm expression product size and purity before digestion. For antibody projects, separate light and heavy chains when possible. For low-abundance targets, plan enrichment or prefractionation before MS rather than after a failed run.
Document sample context before submission. Estimated amount, buffer composition, expected size, disulfide status, glycosylation, and any blocked termini all influence digestion design and interpretation strategy.
Sample Requirements
| Sample Factor | Recommended Condition | Why It Matters |
| Sample format | Purified protein, enriched gel band, or prefractionated material | Reduces spectral complexity and improves target recovery |
| Purity | Single major band or highly enriched target | Lowers contaminant peptides that confuse sequence analysis |
| Protein amount | Enough for repeat digestion and replicate MS when possible | Limited input reduces overlap coverage and repeat analysis |
| Buffer composition | Compatible with digestion; minimal interfering polymers or detergents | Harsh buffers can reduce digestion efficiency and peptide recovery |
| Modifications | Disulfides, glycosylation, phosphorylation, or blocked termini disclosed | Modifications affect digestion, fragmentation, and reporting |
| Background information | Organism, expression system, expected size, or construct sequence if available | Helps choose database-assisted or de novo analysis and set reporting depth |
When sample amount is limited, define realistic coverage expectations before analysis begins. Partial but well-supported regional sequence may still meet the project goal if the required segment is captured with strong peptide evidence.
Step 3: Choose Digestion and Fractionation Strategy
Trypsin is the default protease for many MS-based sequencing workflows, but it is not always optimal. If the target region lacks lysine and arginine sites, complementary proteases such as chymotrypsin, Glu-C, or Lys-C can generate overlapping peptides for assembly. Multiple enzyme digests are especially useful when redundant coverage is required for sequence proof at the protein level.
Digestion conditions should be consistent and documented. Under-digestion creates long peptides that fragment poorly. Over-digestion can destroy informative regions. Reduction and alkylation should be applied when disulfide bonds interfere with solubility or fragmentation. For modified proteins, decide in advance whether modification-aware interpretation is required.
If the sample remains too complex after digestion, use gel sectioning, HPLC prefractionation, or targeted enrichment before MS/MS acquisition. More peptides do not help if spectrum quality drops across a crowded chromatogram.
Step 4: Acquire High-Quality MS/MS Data
Confident protein sequence analysis starts with strong spectra. Use LC separation to reduce precursor overlap. Select precursors with sufficient intensity and informative charge states. Data- dependent acquisition can work well on enriched samples, but complex mixtures may need longer gradients, additional fractionation, or repeat runs.
Practical acquisition priorities include:
Weak spectra with sparse b-ion and y-ion series should not be forced into high-confidence calls. Excluding poor spectra is better than building a sequence conclusion on ambiguous peptide evidence.
Step 5: Perform Sequence Analysis With the Right Logic
Sequence analysis should follow the reporting goal defined at the start. In database-assisted workflows, peptide-spectrum matches are made against a reference proteome or provided construct sequence. This path is efficient when the reference is trustworthy and spectral quality is strong. Unexpected variants, contaminants, or absent entries can still produce partial matches or missed differences.
When no reliable reference exists, de novo peptide interpretation and protein-level assembly become necessary. Analysts derive sequence tags from fragment ions, confirm peptides manually, and align overlaps from one or more digests. Sequence proof at this stage depends on redundant coverage, not on a single strong-looking spectrum.
Manual review remains essential in both paths. Inspect consecutive fragment support, unexplained intense peaks, replicate consistency, and plausible modification assignments. Flag isoleucine/leucine ambiguity and gap regions rather than hiding them in a polished-looking report.
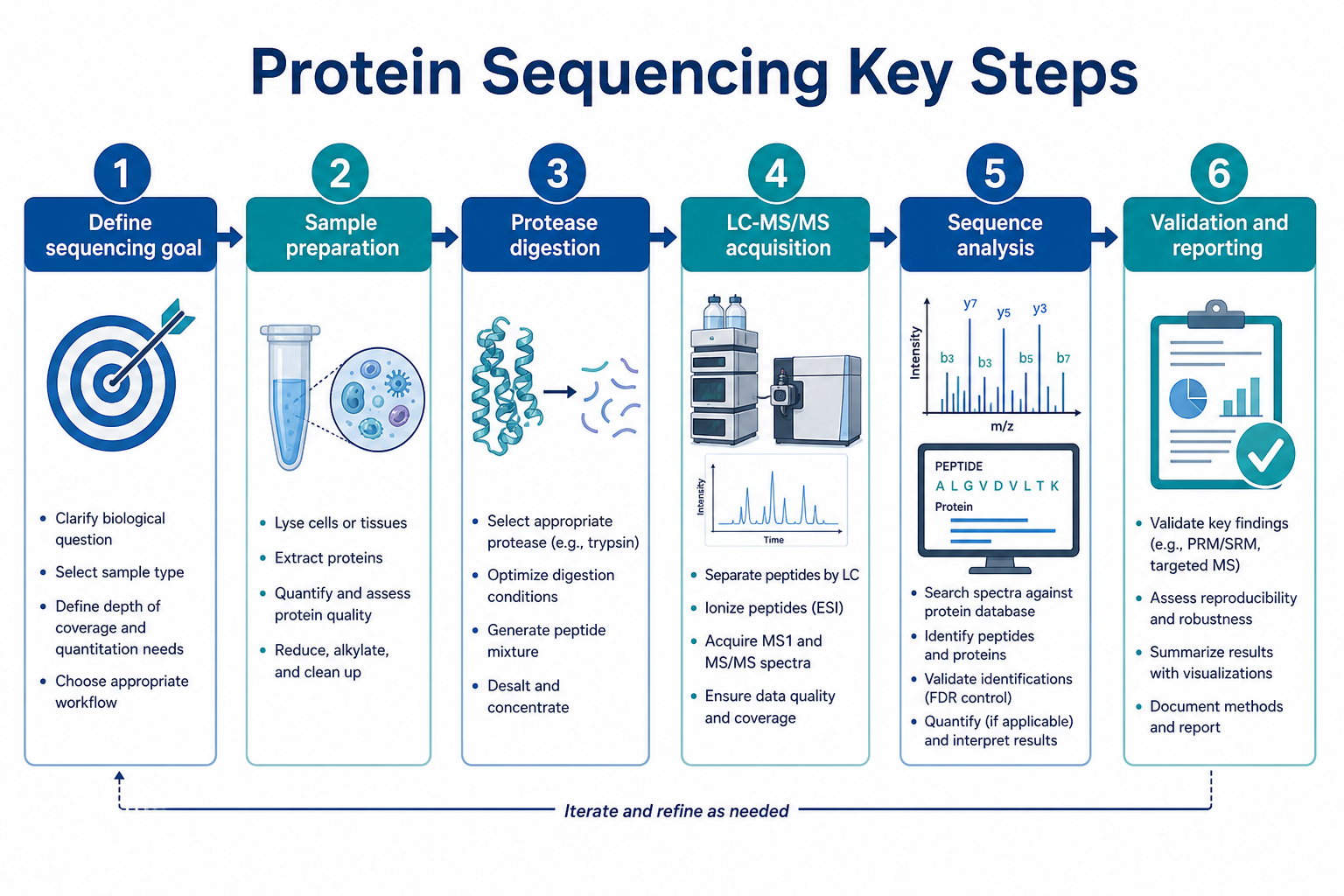
Figure 2. Key steps from project definition through validation and reporting
Sequence analysis should also separate peptide-level findings from protein-level claims. A confirmed peptide supports a local motif. A protein-level sequence map requires overlap logic, gap annotation, and explicit confidence labeling. Mixing these levels in the final report is a common source of downstream dispute.
Step 6: Validate Results and Define Reporting Depth
Validation planning should begin before the final report is written. For high-stakes protein sequencing projects, define which regions require replicate spectra, overlapping peptides, or orthogonal confirmation such as Edman sequencing, gene sequencing, or synthetic peptide standards.
A strong reporting package distinguishes high-confidence segments from tentative calls. It should state where additional digestion, deeper MS, or orthogonal methods would most improve the evidence. Transparent ambiguity labeling is especially important for antibody products, biosimilars, and unknown protein identification.
Expected Outputs From a Well-Run Project
| Output Type | Typical Content | Best Used For |
| Confirmed peptide list | Database-matched or de novo-derived peptides with spectrum support | Regional confirmation and QC review |
| Protein sequence map | Overlapping peptides assembled into a longer sequence | Clone design, homology analysis, construct verification |
| Terminal sequence result | N- or C-terminal readout when terminal method is used | Release testing and terminus verification |
| Annotated spectra | Key MS/MS spectra linked to peptide assignments | Manual audit, publication, or regulatory support |
| Ambiguity flags | I/L positions, low-confidence residues, or gap regions | Transparent reporting and follow-up validation |
| Coverage summary | Region-based or percent coverage of the target protein | Project acceptance and comparability decisions |
The deliverable should match the biological or commercial decision behind the project. Not every study needs full-length coverage, but every study should define what level of evidence is sufficient before analysis begins.
Key Cautions
Do not apply database search when the sample may differ from the reference construct. Do not report full protein sequence from one weak peptide. Do not increase digestion complexity without increasing MS depth. Do not hide manual review when the sequence will support cloning, publication, comparability, or release documentation.
Avoid assuming that more software scores equal more truth. Chimeric spectra, near-isobaric residues, missed cleavages, and partial fragmentation can all produce attractive but incorrect assignments. When material is limited, run a pilot on a small aliquot to test digestion, LC method, and acquisition quality before committing the full sample.
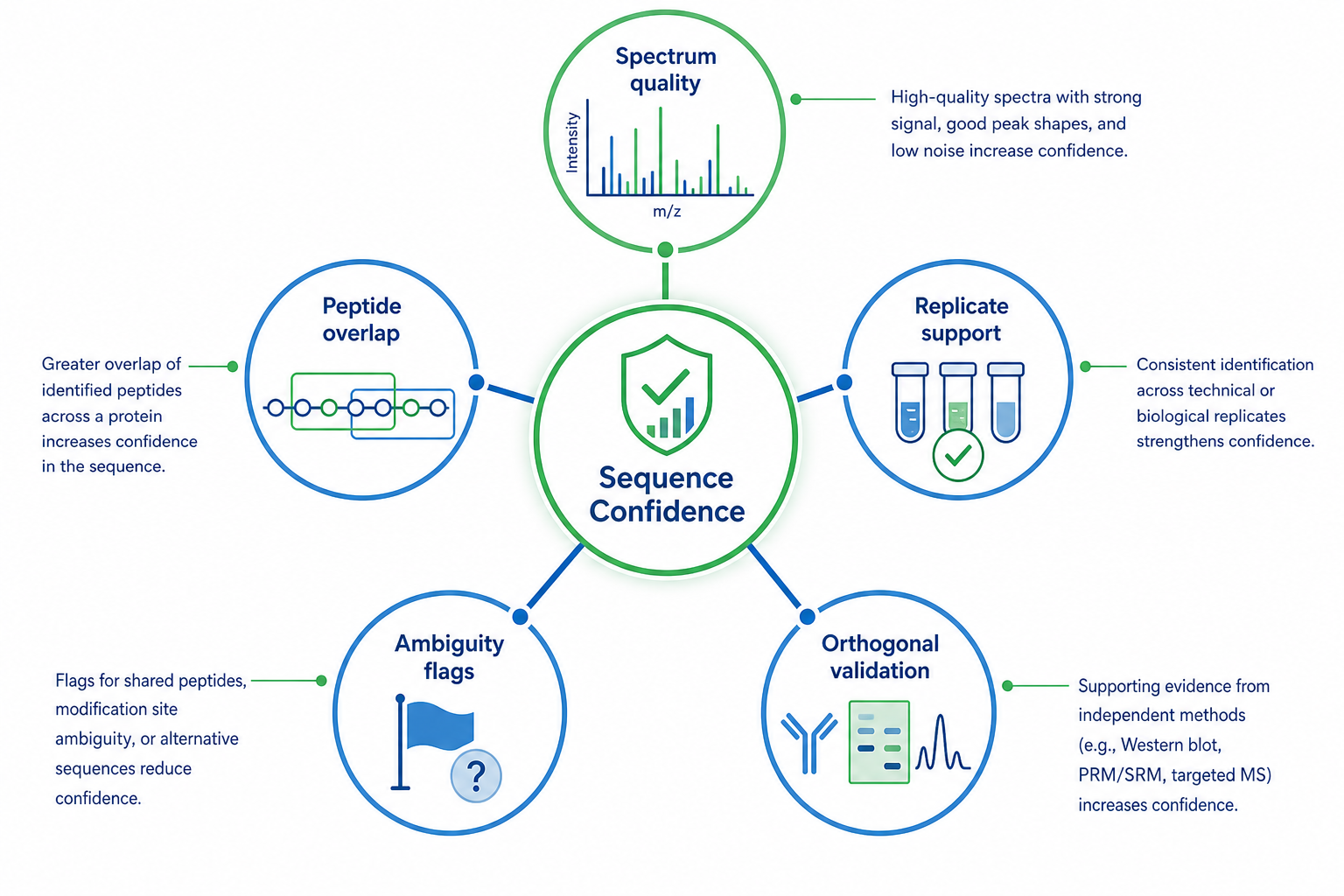
Figure 3. Evidence criteria that support dependable sequence reporting
Pilot testing is especially valuable for antibody chains, membrane proteins, heavily modified biologics, and samples with blocked termini. Early method testing often saves more sample than repeated low-confidence analysis cycles.
Frequently Asked Questions
1. What is the first step in a protein sequence project?
The first step is to define the reporting goal and choose the analysis path. Terminal confirmation, database-assisted tandem MS, and de novo assembly answer different questions and require different sample and MS designs.
2. How much protein is needed?
There is no single amount for every project. Purified or enriched samples with enough material for repeat digestion and replicate MS generally produce stronger overlap coverage. Limited input may still work when the required region is narrow and sample quality is high.
3. When should database search be used?
Database search is appropriate when a trustworthy reference sequence exists and the goal is confirmation or coverage mapping. It is weaker when the protein is unknown, proprietary, truncated, or likely to differ from the reference.
4. What makes protein sequence analysis reliable?
Reliable analysis depends on clean sample input, strong MS/MS spectra, appropriate digestion design, manual spectrum review, overlap support for protein-level claims, and clear reporting of gaps and ambiguous residues.
5. When should researchers outsource MS-based sequence work?
Outsourcing is useful when sample amount is limited, method choice is unclear, spectrum interpretation is uncertain, full-length or antibody coverage is required, or the final protein sequencing report must support cloning, publication, or biopharmaceutical documentation.
Conclusion
Dependable sequence reporting depends on decisions made before and after MS acquisition. Define the reporting goal early, prepare a sequence-friendly sample, choose digestion and acquisition conditions that match the target, perform sequence analysis with the right database or de novo logic, and validate results with transparent reporting standards.
For projects that need MS-based protein sequence confirmation beyond routine identification, contact MtoZ Biolabs to discuss sample prep strategy, LC-MS/MS protein sequencing, terminal sequencing, de novo assembly, or an integrated sequence analysis workflow.
How to order?

