





How to Perform De Novo Protein Sequencing: From Protein Preparation to Sequence Reconstruction
-
a visible protein band is present, but no trustworthy database match exists
-
peptide coverage is too low to support full-length assembly
-
multiple co-migrating proteins reduce sequence confidence
-
harsh buffers or low sample amount limit digestion quality
-
repetitive or modified regions create ambiguous assembly
-
the project requires a sequence report suitable for cloning, QC, or publication, not just tentative peptide IDs
-
Is terminal confirmation enough, or is full-length sequence reconstruction required?
-
Is the protein truly unknown, or is the goal to verify an uncertain reference?
-
Will the report support cloning, publication, internal QC, or tech transfer?
-
Is partial coverage acceptable if unsupported regions are clearly documented?
-
obtain high-quality MS/MS spectra across the peptide mixture
-
maximize peptide detection without overloading the LC column
-
acquire enough instrument depth to support low-abundance peptides when needed
-
document replicate injections when reproducibility must be demonstrated
-
prioritize high-confidence spectra with clear fragmentation ladders
-
resolve ambiguities caused by isobaric residues when possible
-
account for modifications that shift peptide mass or fragment pattern
-
avoid forcing assembly across weak or unsupported regions
-
overlapping peptides from one or more digests
-
consistent alignment across repeat LC-MS/MS runs or replicates
-
clear coverage of N-terminal and C-terminal regions when full-length reporting is required
-
documented gaps or low-confidence regions rather than silent omission
-
N-terminal or C-terminal sequencing for end confirmation
-
intact mass measurement for global mass consistency
-
peptide mapping against a provisional sequence when a reference becomes available
-
repeat digestion with an alternative protease to close coverage gaps
Introduction
De novo protein sequencing is often requested when a purified protein must be characterized, but no reliable reference sequence is available. A gel band from an expression experiment, a legacy purified fraction, or a recombinant batch with incomplete records can all create the same problem. The protein is present, yet the amino acid sequence remains unknown. Database-assisted identification may return weak matches, partial coverage, or no usable assignment. In these cases, the project needs sequence evidence derived directly from the sample.
Related Services
| Service Area | Recommended Service |
|---|---|
| De novo protein sequencing | |
| Full-length protein sequencing | |
| Protein identification | |
| Terminal confirmation | / |
| Sample preparation support | |
| Unknown protein sequencing |
Teams preparing a first de novo protein sequencing submission can consult MtoZ Biolabs to review sample purity, digestion strategy, and expected coverage before LC-MS/MS analysis begins.
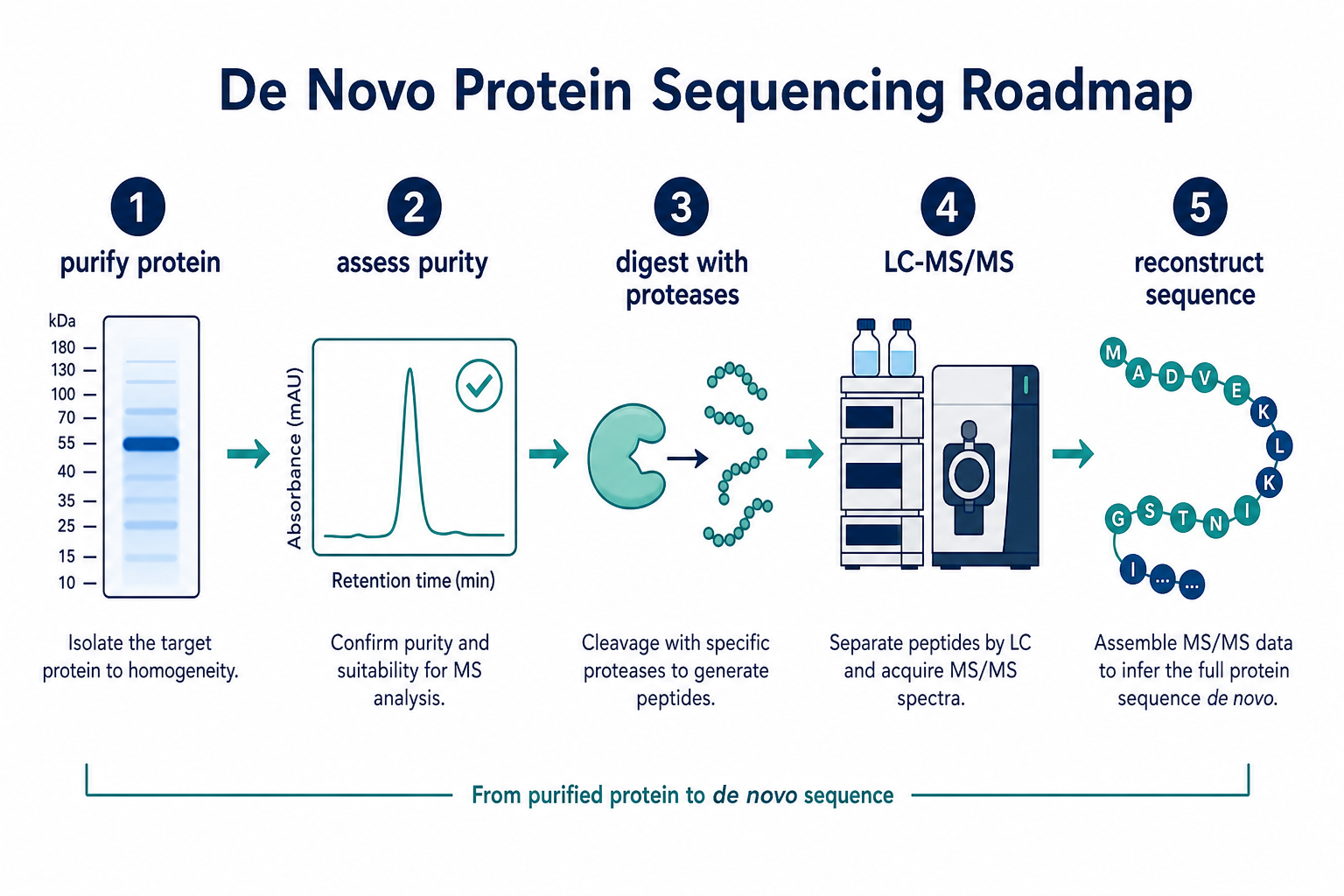
Figure 1. De novo protein sequencing moves from purified protein through digestion, LC-MS/MS, and sequence reconstruction.
Common Pain Points Before Starting
Researchers often begin a de novo protein sequencing project after encountering one or more of these problems:
These issues are common for unknown proteins, proprietary constructs, enriched fractions, and recombinant products without complete genetic records. The practical question is not whether sequencing is theoretically possible. The question is whether the sample and workflow can generate enough overlapping peptide evidence for a defensible sequence.
Why De Novo Sequencing Projects Fail Early
Most early failures come from preparation and design rather than from spectral interpretation alone.
Insufficient purity. Mixed proteins produce competing peptide signals and make assembly unreliable.
Incompatible sample matrix. High salt, strong detergents, chaotropic agents, and some storage buffers interfere with digestion or LC-MS/MS.
Single-protease limitation. One enzyme may leave long regions without useful peptides, especially across hydrophobic or modified domains.
Weak MS/MS quality. Low signal, poor fragmentation, or limited instrument time reduces the number of interpretable spectra.
Unrealistic coverage expectations. Long, modified, or repetitive proteins may need multiple digests, repeat LC-MS/MS runs, and orthogonal confirmation before full-length reconstruction is possible.
Understanding these root causes helps researchers fix the workflow before resubmitting material.
Step 1: Define the Project Goal and Deliverable
Before sample preparation begins, define what the project must deliver.
A feasibility review should match method depth to project need. A terminal check requires a different workflow than full-length de novo assembly.
Step 2: Prepare and Assess the Protein Sample
Sample quality is the highest-leverage step in de novo protein sequencing.
1. Confirm Purity
Review SDS-PAGE, staining pattern, and purification history. A dominant band does not always mean a single protein. Excise gel bands precisely and avoid contamination from neighboring lanes or higher-abundance proteins.
2. Check Integrity and Amount
Confirm that enough material is available for digestion, cleanup, and repeat LC-MS/MS if needed. Degraded or partially proteolysed protein reduces usable peptide evidence.
3. Document Sample History
Record expression system, expected molecular weight, purification method, storage conditions, and any known modifications. This information helps interpret unexpected truncations or processing events later.
4. Clean Up Incompatible Buffers
Exchange or remove salts, detergents, and interfering additives when possible. In-solution digestion works best with protein in a digestion-compatible buffer after appropriate cleanup.
Step 3: Choose the Digestion Strategy
Digestion design determines peptide coverage across the protein backbone.
Trypsin is the most common protease because it produces peptides suited to LC-MS/MS. However, a single protease may not cleave evenly across the entire sequence. Complementary enzymes such as chymotrypsin, Glu-C, Lys-C, or Asp-N can improve overlap in difficult regions.
| Digestion Approach | Best Use Case | Main Advantage | Main Risk |
|---|---|---|---|
| In-solution trypsin digestion | Purified soluble protein | Efficient workflow for clean samples | Buffer interference if cleanup is skipped |
| In-gel digestion | SDS-PAGE band or low-purity sample | Removes detergents and focuses on target band | Band contamination reduces confidence |
| Multi-enzyme digestion | Full-length reconstruction | Broader peptide overlap | More complex analysis and sample demand |
| Targeted repeat digestion | Coverage gaps after first run | Can rescue unsupported regions | Requires additional material and time |
For unknown proteins, multi-enzyme digestion is often the most practical route to sequence reconstruction.
Step 4: Perform LC-MS/MS Acquisition
After digestion, peptides are separated by liquid chromatography and analyzed by tandem mass spectrometry. High-resolution MS/MS spectra provide the fragmentation patterns used for de novo interpretation.
Key acquisition goals include:
Weak spectra are a common bottleneck. If initial runs produce sparse fragmentation, additional LC-MS/MS time, fractionation, or repeat analysis may be required before sequence reconstruction can proceed.
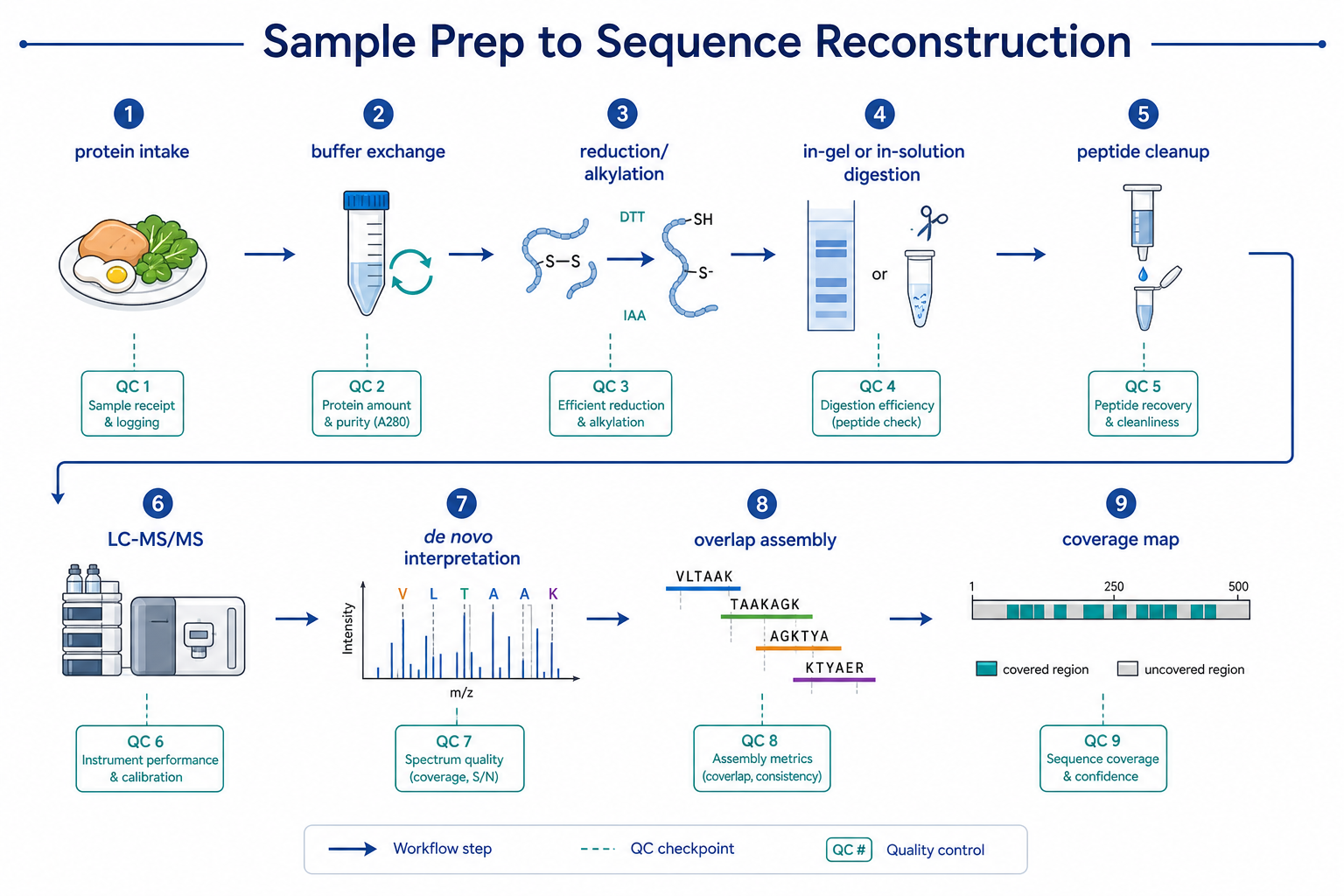
Figure 2. A complete de novo workflow includes sample cleanup, digestion, peptide analysis, spectral interpretation, overlap assembly, and coverage mapping.
Step 5: Interpret Spectra Without a Trusted Reference
De novo interpretation derives amino acid sequence information directly from peptide fragmentation patterns. Analysts use b-ion and y-ion series to build short sequence tags from individual MS/MS spectra.
Important interpretation principles include:
Database search may still be used as a supporting tool, but de novo protein sequencing should not depend on a reference that is itself uncertain.
Step 6: Reconstruct the Protein Sequence
Sequence reconstruction aligns overlapping peptides into longer contiguous regions. The goal is to build a protein-level sequence supported by multiple independent peptide matches.
A strong reconstruction usually shows:
For long proteins, reconstruction may proceed in blocks. Unsupported segments should be flagged explicitly so the report matches the evidence level.
Step 7: Validate and Report the Final Sequence
The final report should state what is supported, what is inferred, and what remains unverified.
Useful validation options include:
A publication-ready or QC-ready report should include coverage maps, confidence notes, and any limitations caused by modifications, homology, or sample constraints.
Expected Results and How to Judge Success
A successful de novo protein sequencing project may deliver one of three outcomes depending on the goal.
Terminal or partial sequence confirmation. Useful when only specific regions must be verified.
High-confidence partial assembly. Useful when most of the sequence is needed for cloning or internal decision-making, with known gaps documented.
Full-length or near-full-length reconstruction. Useful when the project requires broad primary structure evidence from purified protein material.
Success should be judged by evidence quality, not by peptide count alone. A smaller set of well-supported overlapping peptides is more valuable than a large list of weak identifications.
Troubleshooting Common Problems
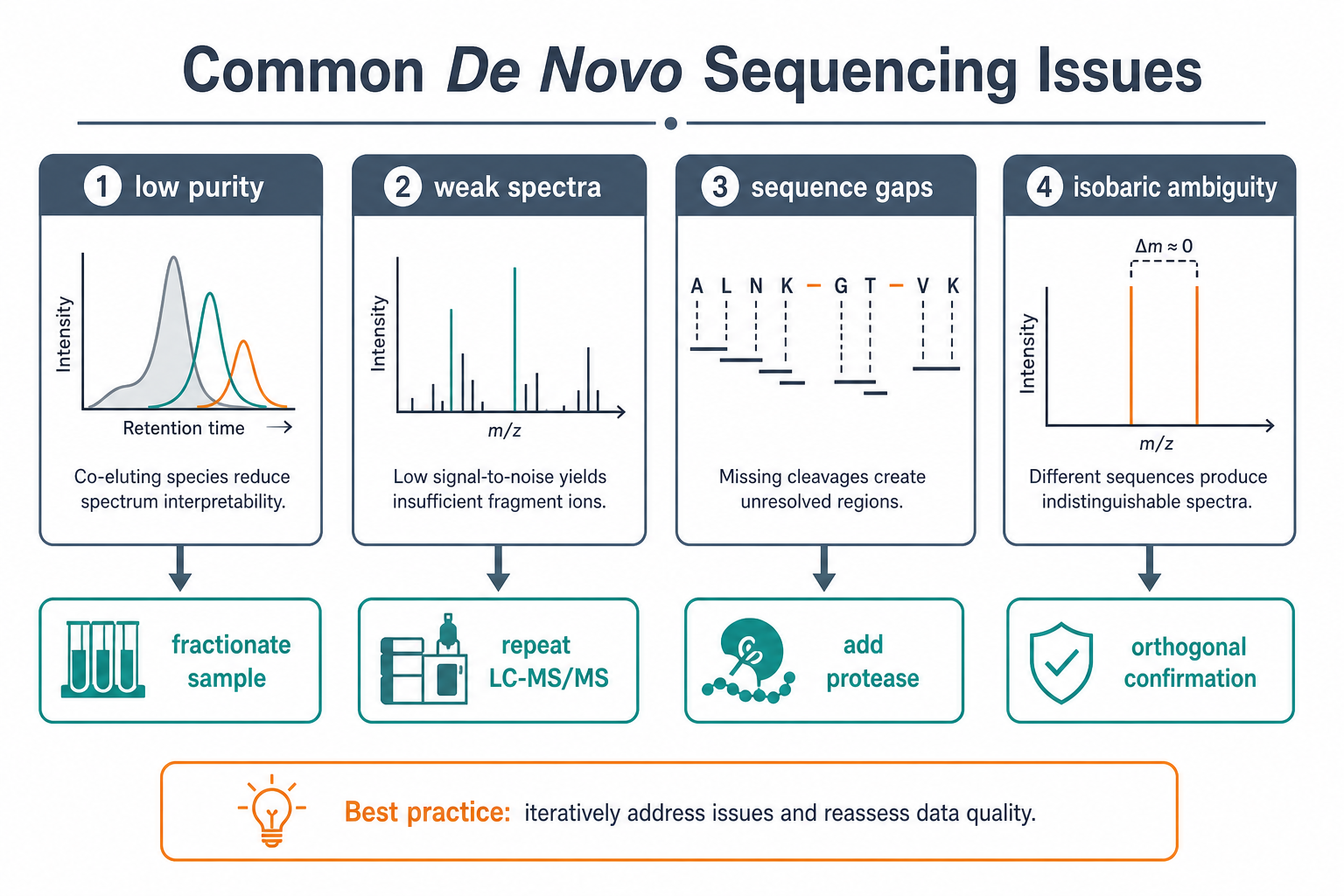
Figure 3. Low purity, weak spectra, sequence gaps, and isobaric ambiguity are common issues that can often be addressed by workflow adjustment.
| Problem | Likely Cause | Recommended Fix |
|---|---|---|
| Low peptide yield | Sample amount, degradation, or buffer interference | Clean up sample, increase input, repeat digestion |
| Weak MS/MS spectra | Low abundance peptides or instrument depth limits | Increase LC-MS/MS time, fractionate sample, repeat run |
| Coverage gaps | Single-protease bias or resistant domains | Add complementary protease, redesign digestion |
| Mixed sequence evidence | Co-purified proteins or gel contamination | Improve purification or excise band more precisely |
| Ambiguous residues | Isobaric amino acids or homologous regions | Use orthogonal confirmation and expert manual review |
If troubleshooting does not improve coverage, the project may need a staged plan with partial sequence delivery and targeted follow-up rather than repeated identical submissions.
Key Precautions
Do not assume that a clean gel band guarantees successful de novo protein sequencing. Purity, amount, and buffer compatibility still matter.
Do not rely on one protease when full-length reconstruction is required. Coverage gaps are common with single-enzyme workflows.
Do not treat tentative peptide tags as a finished protein sequence. Assembly and expert review are essential.
Do not skip documentation of unsupported regions. A transparent report is more useful than an overconfident partial sequence.
For rare or difficult samples, a pilot feasibility review can prevent unnecessary loss of material.
Frequently Asked Questions
1. How much purified protein is needed for de novo protein sequencing?
Requirements vary by protein length, purity, and coverage goal. Terminal or partial sequencing may need less material than full-length reconstruction. A feasibility review before submission is recommended.
2. Can de novo protein sequencing work from an SDS-PAGE band?
Yes. In-gel digestion is a common route when the target protein is separated by gel electrophoresis. Band purity and precise excision strongly affect the result.
3. Why is multi-enzyme digestion often used?
Different proteases cleave at different sites and produce overlapping peptides. This overlap improves the chance of reconstructing longer contiguous sequence regions.
4. Can de novo sequencing distinguish leucine and isoleucine?
Routine workflows often cannot distinguish these isobaric residues by mass alone. Project reports should note this limitation when relevant.
5. When should terminal sequencing be added?
Terminal sequencing is useful when the project requires confirmation of the N-terminus or C-terminus, or when full-length coverage is strong but end validation is still needed for the final report.
Conclusion
De novo protein sequencing moves from purified protein preparation through digestion, LC-MS/MS, spectral interpretation, and sequence reconstruction. Strong results depend on defining the deliverable early, preparing a clean sample, designing digestion for coverage, acquiring high-quality MS/MS data, and reporting supported sequence regions transparently. When the reference is missing or unreliable, this workflow provides direct primary structure evidence from the protein itself. Researchers preparing unknown protein, recombinant, or legacy samples can contact MtoZ Biolabs to review sample readiness and build a de novo protein sequencing plan from preparation through final sequence reconstruction.
How to order?

