





How to Interpret PhIP-Seq Results for Autoantibody Discovery and Candidate Validation
- single enriched peptide
- cluster of overlapping peptides
- mapped antigen candidate
- cohort-defined subgroup signal
- candidates found in multiple case samples with low control frequency
- subgroup-specific enriched peptides linked to a plausible study hypothesis
- antigen-region patterns repeated across independent samples
- candidates that remain interpretable after motif and control review
An enriched peptide in PhIP-Seq should move forward only when the signal stays above background, lines up with related evidence, and points to a clear next experiment. In autoantibody discovery, the strongest candidates usually combine reproducible peptide enrichment, low background binding in negative control or beads-only control samples, coherent overlapping peptides within one antigen region, and a case-associated pattern that can guide orthogonal validation or cohort stratification.
In practice, phip seq data analysis is a triage problem, not a hunt for the single tallest peak. Most teams need to review four linked levels at once: peptide-level signal, epitope mapping, antigen-level interpretation, and cohort stratification. That framework helps decide which findings justify peptide ELISA, peptide array, protein array, immunoblot, or antigen-specific immunoprecipitation, and which ones should stay exploratory.
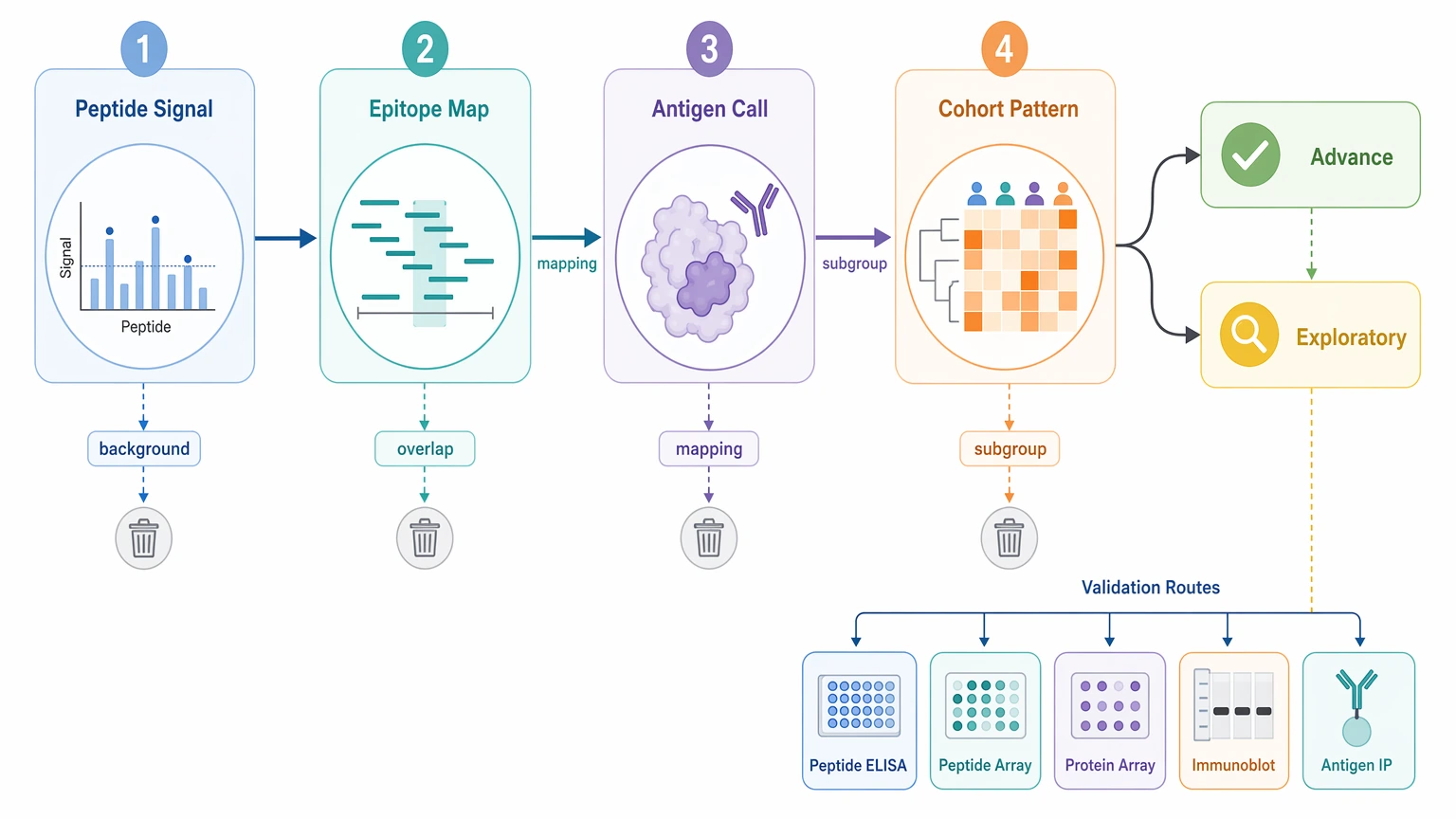
Where Interpretation Usually Gets Stuck
The hard part begins after enrichment calling, when many hits look usable but only a few can justify more sample volume, assay development, and validation budget. One sample may show a strongly enriched peptide with no neighboring support. Another candidate may show several overlapping peptides across the same protein region. A third pattern may come from a short sequence motif repeated across unrelated proteins. Some signals also show up in both cases and controls, just at different frequencies.
Those outcomes do not mean the same thing. Advancing weak hits too early can send effort toward background-prone sequences or ambiguous mappings. Filtering too hard can remove subgroup-restricted signals that matter for follow-up assay design or cohort stratification. The real question is not whether a peptide enriched, but whether the overall pattern is strong enough to support candidate validation.
What the Output Actually Represents
PhIP-Seq, or phage immunoprecipitation sequencing, measures antibody binding to a phage-displayed peptide library, followed by immunoprecipitation and next-generation sequencing. The output is peptide enrichment. It is not direct proof that antibodies bind the native full-length antigen, and it does not establish recognition of a conformational epitope.
That distinction shapes every downstream decision. A strong peptide-level signal may reflect a true linear epitope, but it may also come from library representation bias, shared sequence motif behavior, or nonspecific pull-down. Good interpretation keeps those possibilities in view instead of collapsing everything into one protein name too soon.
Brief Root-Cause Analysis of False Positive Prioritization
1. Overcalling isolated enriched peptides
A single enriched peptide can look convincing in fold-enrichment or z-score terms and still be biologically unstable. Without overlapping peptides, replicate concordance, or a useful case-control pattern, the hit may reflect selection noise rather than a durable autoantibody target.
2. Ignoring sequence motif and mapping ambiguity
Some enriched peptides contain low-complexity segments or short motifs shared across several proteins, domains, or isoforms. In those cases, antigen-level interpretation is less certain than the top-ranked parent annotation suggests.
3. Ranking hits mainly by magnitude
Large counts can flag candidates, but count magnitude alone is a weak prioritization rule. A moderate peptide-level signal repeated across multiple case samples and absent from negative control samples often deserves more attention than an extreme one-sample event.
4. Choosing the wrong validation format
A narrow peptide-region discovery does not automatically call for a full-length protein assay. If the data point to a linear epitope, peptide-based follow-up may test the finding more directly.
Related Services
Main Service |
Supporting Service |
Validation Service |
Alternative Service |
A Step-by-Step Framework for Data Interpretation
Step 1: Confirm that the peptide-level signal is technically credible
Start with the normalized metric used in your pipeline, whether that is fold-enrichment, z-score, RPM-style normalization, rpK, or a model-based enrichment statistic. Then ask the practical question: does the enriched peptide separate from input, mock, negative control, or beads-only control samples? A candidate worth advancing should separate from background, not just post a large absolute count.
Replicate concordance belongs in the same review. If you have repeated serum runs, technical repeats, or split batches, compare both enrichment rank and direction of effect. A peptide that rises once and then collapses on repeat should fall off the shortlist.
Step 2: Define the interpretation unit before ranking hits
Do not put every result into one flat hit table. First sort findings into four units:
This simple split improves prioritization. A single peptide event and a multi-peptide antigen pattern may both count as enriched, but they do not offer the same validation readiness.
Step 3: Look for overlapping peptides and epitope continuity
Overlapping peptides often provide the strongest internal support for a real linear epitope. When several adjacent tiled peptides enrich across the same antigen region, the pattern supports epitope continuity rather than random capture. It still does not prove native full-length recognition, but it gives the team a more defensible candidate and a clearer basis for assay design.
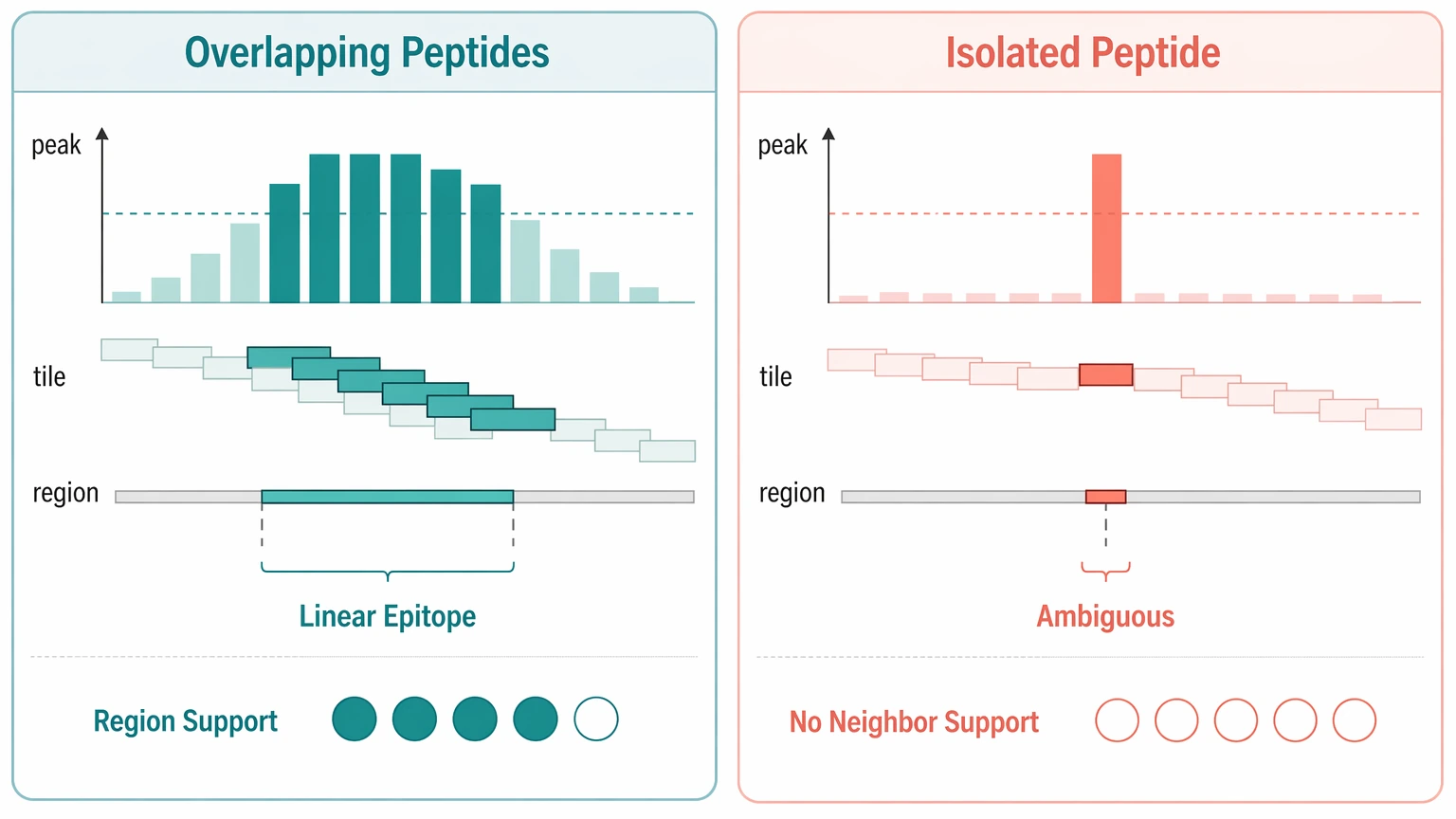
An isolated spike with no neighboring support deserves a tougher review. Check for local tiling gaps, abundance effects in the phage-displayed peptide library, recurring sequence motif behavior, and whether the region is poorly represented. In most autoantibody discovery projects, region-level continuity is more actionable than one sharp peak alone.
Step 4: Test whether antigen-level interpretation is clean enough to act on
After peptide mapping, ask whether the parent assignment is unique and biologically interpretable. Some enriched peptides map to homologous proteins, repeated domains, or multiple isoforms. When that happens, the protein name can imply more certainty than the data support. Keep the region or motif annotation visible in the shortlist so downstream planning stays accurate.
Library representation matters here too. If a protein has sparse coverage, the absence of adjacent enriched peptides is less informative. If a well-covered antigen shows only one enriched peptide across a densely tiled region, skepticism is more appropriate.
Step 5: Use cohort distribution to decide follow-up value
For candidate validation, ask two direct questions: does the signal separate cases from controls, and does it define a meaningful subgroup? Some autoantibody candidates are not shared across all cases. That does not automatically weaken them. A subgroup-restricted pattern can still support cohort stratification if it is reproducible and biologically interpretable.
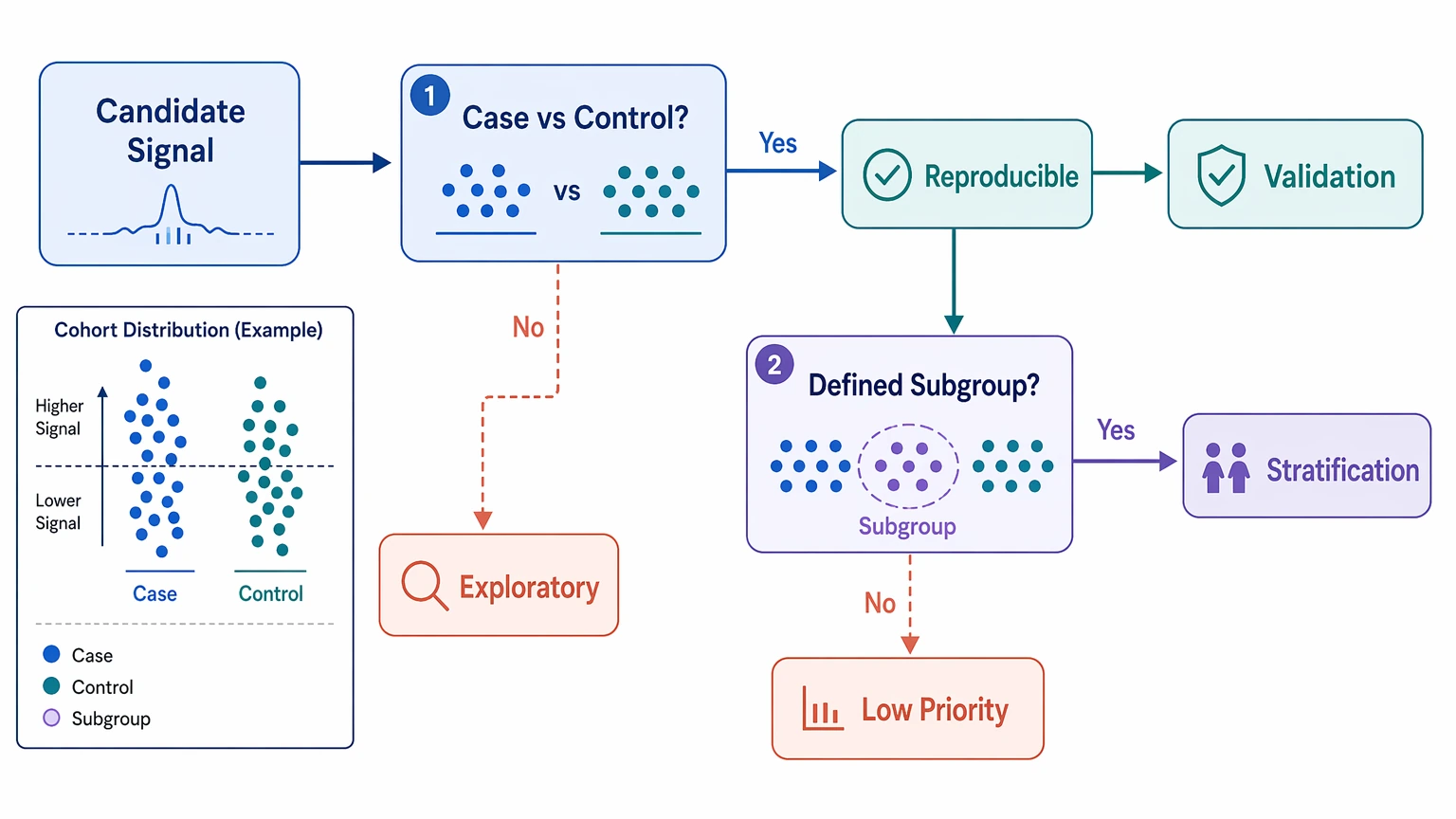
A practical shortlist often includes:
If your team needs a structured review of peptide enrichment, background binding, and orthogonal validation fit, MtoZ Biolabs can evaluate your project workflow; submit your requirements to discuss which candidates are ready for follow-up testing.
Step 6: Match each candidate to the right orthogonal validation route
The confirmatory assay should reflect what PhIP-Seq actually measured. If the evidence centers on a short linear epitope, peptide-based validation is often the most direct next step. If the pattern suggests a broader region, or if native folding may influence antibody binding, a domain-based or full-length protein format may be more informative.
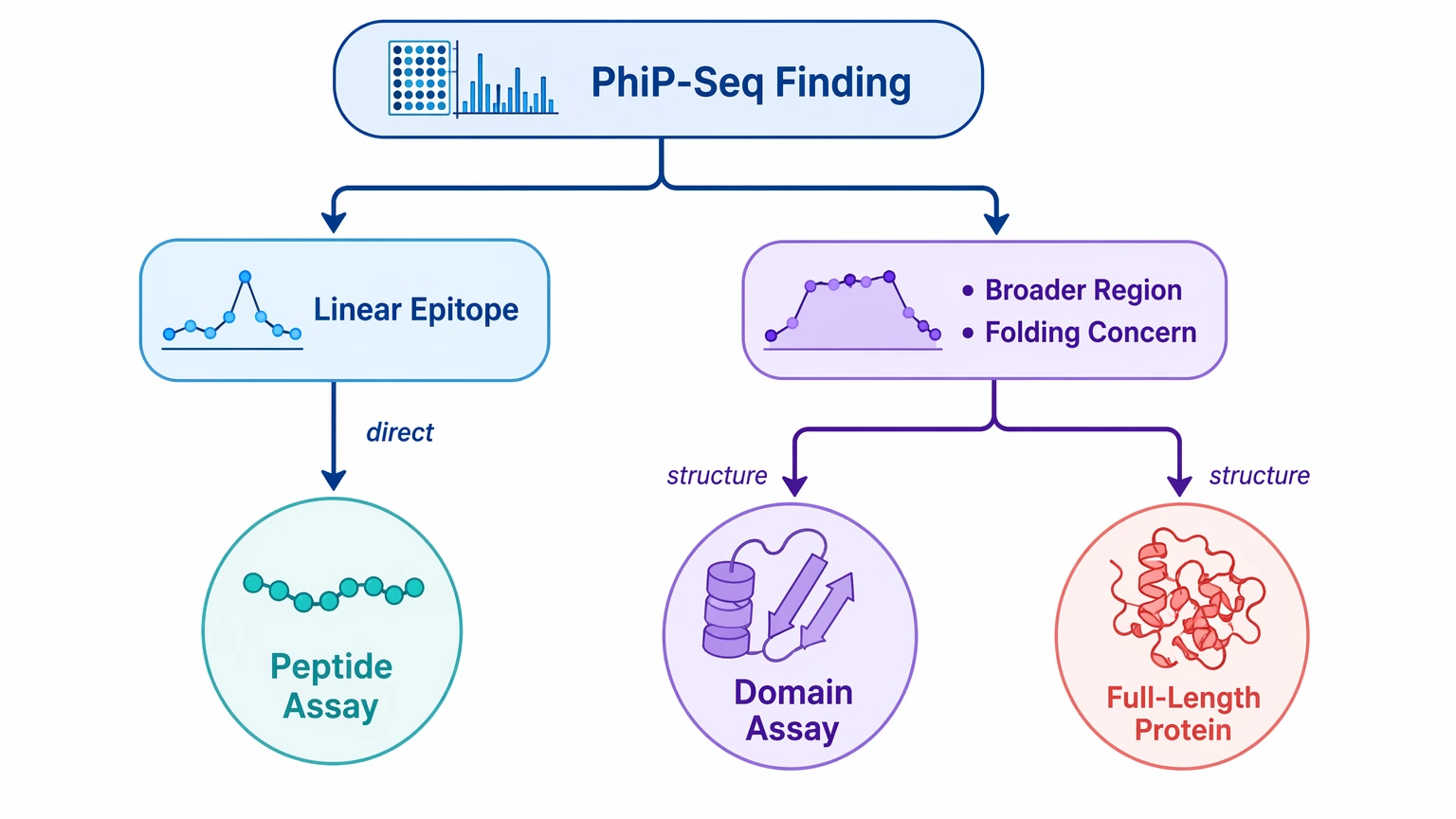
This step avoids a common misread: testing a narrow peptide discovery only with a full-length antigen assay and then treating a negative result as disproof. In some studies, the better path is peptide-first confirmation, followed by protein-level testing only when the biology supports that move.
Common Interpretation Mistakes to Avoid
Several mistakes repeatedly distort phage immunoprecipitation sequencing interpretation. One is treating PhIP-Seq as fully quantitative in the same sense as a calibrated immunoassay. Another is assuming every enriched peptide maps cleanly to one actionable antigen. A third is skipping control review because a cohort pattern looks interesting. Teams also overstate discovery-stage disease association when the current evidence only supports hit prioritization.
Keep the platform limits in view. PhIP-Seq is especially useful for linear epitope discovery across large peptide spaces, but conformational epitope reactivity may be underrepresented. A negative result does not rule out antibody binding to a native folded target.
Conclusion
The most useful PhIP-Seq candidates are not simply the highest sequencing peaks. They are the signals that stay above background, show replicate concordance, form coherent overlapping peptide patterns when possible, support careful antigen-level interpretation, and offer real value for cohort stratification or follow-up assay design. That approach fits discovery-stage projects that need a defensible shortlist rather than a broad inventory of enriched peptides.
For projects with ambiguous peptide clusters, motif-sharing concerns, or uncertainty about peptide versus protein follow-up, contact MtoZ Biolabs to evaluate your project and align the PhIP-Seq readout with an appropriate candidate validation plan before committing additional samples or assay development effort.
FAQ
Should I aggregate peptide-level signal to one antigen score before review?
Usually not at the start. Review peptide-level signal, overlapping peptides, and mapping ambiguity first. Early aggregation can hide whether the apparent antigen call comes from one peptide, one motif, or a coherent region.
What should I do when controls are limited or uneven across batches?
Treat borderline candidates conservatively. In that setting, replicate concordance, internal overlap patterns, and within-batch case-control comparisons matter more than cross-batch rank order. If uncertainty remains high, a rerun is often more informative than forced progression.
How should I handle enriched peptides shared across homologous proteins?
Keep the call at the motif or region level until an orthogonal assay can separate the targets. The signal may still be relevant, but it is not yet a clean antigen-specific assignment.
Can a candidate move forward without overlapping peptides if library coverage is sparse?
Yes. Sparse library representation changes how much weight you can place on missing neighbors. In that situation, repeatability, case-associated frequency, and a validation assay built around the exact displayed region become more important.
When does cohort stratification matter more than overall case frequency?
It matters when the project goal is to identify biologically distinct subgroups rather than a broadly shared serologic marker. A lower-frequency signal can still justify follow-up if it clusters within a defined phenotype, treatment history, or mechanistic subgroup.
What is the best first confirmatory assay when conformational binding is a concern?
Move beyond peptide-only formats early. If native structure may influence antibody binding, test a domain-based or full-length protein assay alongside, or immediately after, peptide confirmation.
How to order?

