





How to Improve Identify Unknown Autoantibodies Optimization in Research Workflows
- Unknown autoantibodies may recognize conformational antigen features that are lost on denatured proteins or short peptides.
- Post-translational modification may define binding, so an unmodified recombinant target or a standard array feature may miss the relevant epitope.
- Immune complex enrichment is selective, and different enrichment conditions change which targets are recoverable for downstream mass spectrometry.
- whether the same collection type is used across all discovery and control samples
- whether samples were processed under similar timing conditions
- whether input amount remains sufficient after depletion or enrichment steps
- whether negative controls show stable background binding
- Protein microarray formats can support broad screening when the goal is to compare many full-length or near-full-length targets, but performance depends on how proteins were produced, folded, and immobilized.
- Peptide array formats are useful when linear epitopes are suspected or when teams want to refine narrowed candidates, but they may miss conformational antigen recognition.
- Immunoprecipitation can preserve more native interactions and is often helpful when the target may exist in a complex or requires a particular cellular context.
- Mass spectrometry becomes most informative after the enrichment strategy and background controls are strong enough to distinguish co-purified noise from plausible target candidates.
- stronger separation between sample and negative-control enrichments
- better replicate overlap among shortlisted proteins
- fewer one-off identifications with no technical or biological support
- improved peptide or protein identification confidence under the same filtering rules
- reproducible detection across technical repeats
- signal-to-noise improvement relative to controls
- persistence across an orthogonal method
- compatibility with the suspected antigen class or biological compartment
- plausible epitope presentation in the assay used
- follow-up feasibility for target confirmation
- confirming a protein microarray hit with immunoprecipitation followed by targeted detection
- testing immunoprecipitation-mass spectrometry candidates on a peptide array to examine support for a linear epitope
- comparing recombinant target binding with a more native source material when a conformational antigen is suspected
- evaluating whether modified versus unmodified antigen changes the binding pattern when post-translational modification may matter
- overlap of top-ranked candidates across methods
- lower background binding rate in controls
- reduced batch-to-batch variance after optimization
- stable confirmation trends across independent sample aliquots
- a manageable number of retained candidates after filtering
- repeated low-confidence hits despite multiple assay rounds
- disagreement between screening and target confirmation methods
- limited remaining sample and little room for broad trial and error
- need for immunoprecipitation and mass spectrometry integration with tighter control design
- uncertainty about whether observed reactivity reflects true antigen specificity, co-enriched complexes, or non-specific binding
Quick Answer: How to Improve an Unknown Autoantibody Identification Workflow
A research team can improve an unknown autoantibody identification workflow by tightening four decision points in sequence: sample quality control, antigen presentation strategy, discovery assay fit, and orthogonal target confirmation. When results are weak, non-specific, inconsistent across runs, or hard to validate, the first step is to identify where confidence drops. In most exploratory research workflows, the main limitation is not one failed assay. It is a mismatch among sample biology, epitope presentation, enrichment design, and the confirmation method.
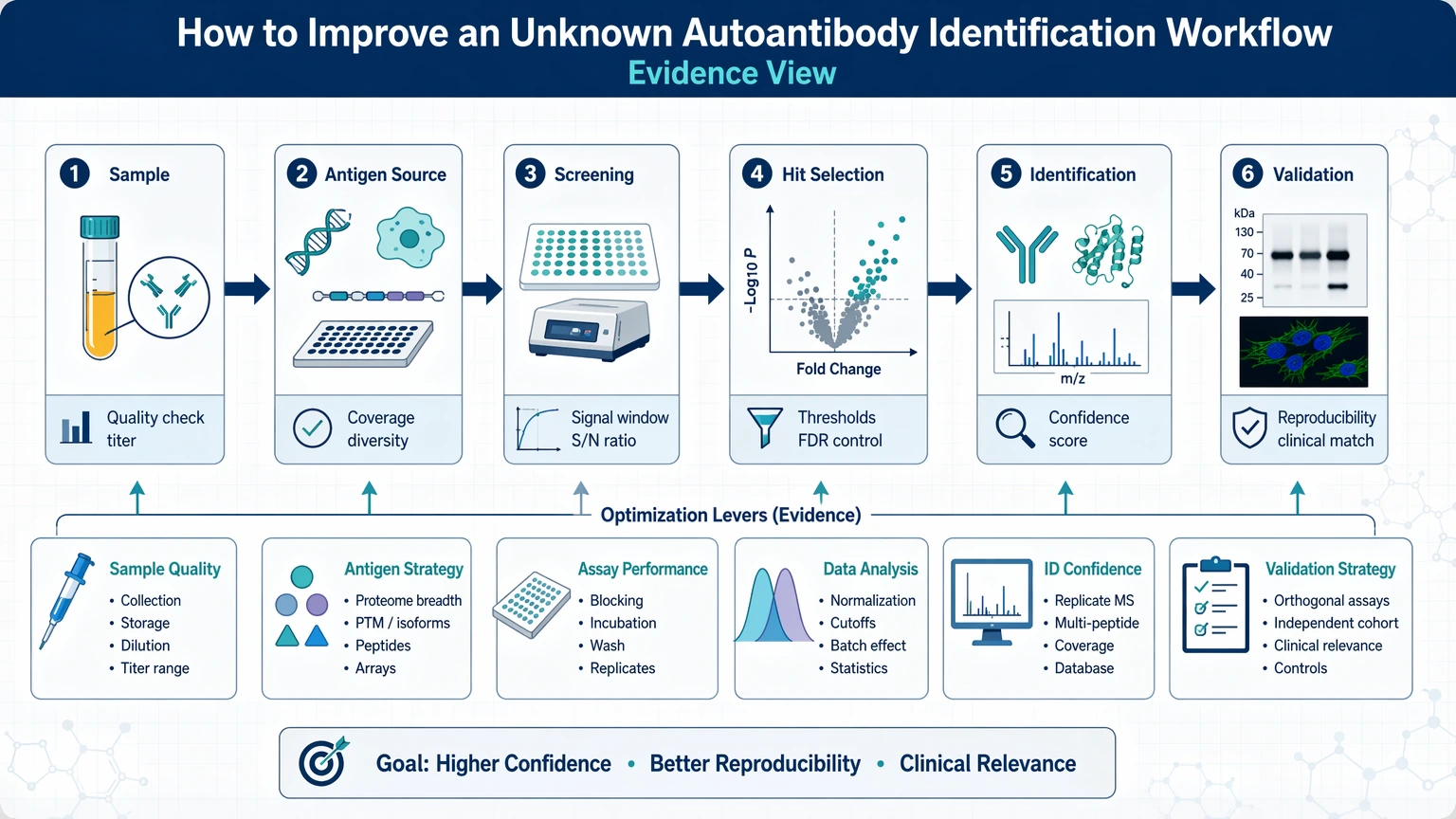
A practical decision path looks like this:
1. Verify that the sample still contains detectable antibody activity after storage, freeze-thaw exposure, depletion, or pretreatment. 2. Match the screening format to the suspected antigen class instead of assuming one platform will capture all unknown autoantibodies. 3. Control non-specific binding before expanding the candidate list, especially when serum or plasma matrix effects are likely. 4. Use orthogonal validation early to test whether top hits remain credible under a different assay mechanism. 5. Rank candidates with predefined filters such as replicate consistency, background separation, enrichment trend, and feasibility of target confirmation.
If broad background, poor replicate agreement, or weak overlap between screening and follow-up assays persists after these adjustments, the workflow usually needs redesign rather than more repetition.
Why Unknown Autoantibody Identification Often Stalls in Research
Unknown autoantibodies are hard to resolve because the signal depends on two variables at once: the antibody population in the sample and the way antigens are presented during screening. That uncertainty starts before data analysis.
In exploratory and translational research, teams often begin with a real biological observation such as tissue reactivity, unusual immunoblot bands, or unexplained enrichment patterns. The challenge is converting that observation into defensible antigen specificity. Workflows stall when an initial assay detects binding activity, but the next step cannot separate true target engagement from matrix interference, cross-reactivity, or incomplete antigen representation.
Three technical realities explain many bottlenecks:
For that reason, identify unknown autoantibodies optimization is less about finding one stronger assay and more about aligning workflow design with the form of antigen the sample is most likely to recognize.
Common Failure Signals in Autoantibody Discovery Workflows
Most underperforming workflows show recognizable failure patterns. Defining the pattern first helps narrow the likely cause.
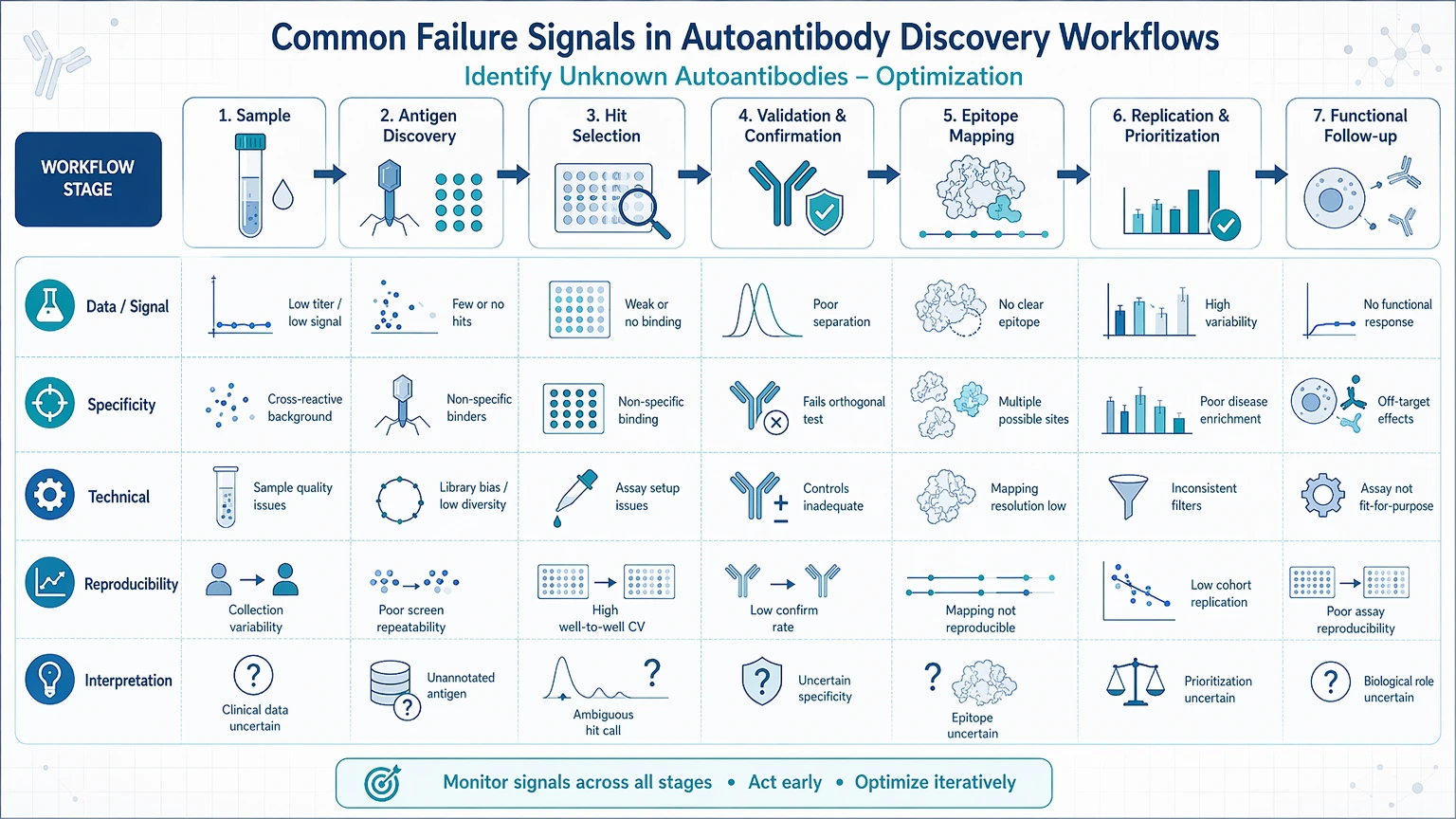
Weak signal with no clear ranking
This pattern usually means the assay detects some binding but does not create enough separation between likely positives and background. Common causes include low antibody abundance, over-dilution, sample degradation, low-efficiency immune complex enrichment, or antigen density that is too low to support stable interaction.
Broad background across many antigens
When many features score above baseline, the problem is often non-specific binding rather than broad biological reactivity. Serum or plasma matrix effects, incomplete blocking, sticky immunoglobulin fractions, rheumatoid factor-like interference, or overly permissive hit thresholds can all inflate the candidate list.
Discovery hits fail in confirmation assays
This is one of the most informative failure modes. If a protein microarray or immunoprecipitation experiment produces attractive candidates but follow-up assays do not confirm them, ask whether the original hit depended on assay-specific presentation, denatured versus native structure, shared motifs, or co-enriched binding partners rather than direct antigen specificity.
Poor reproducibility across runs or operators
Inconsistent results across batches often reflect variable sample handling, shifting wash stringency, unstable antigen lots, inconsistent normalization, or low signal-to-noise near the assay detection floor. Repeating the same workflow without controlling these variables usually adds noise rather than confidence.
Too many low-confidence candidates
A long candidate list is not always a discovery advantage. It often signals weak prioritization logic. If the workflow does not define filters for replicate overlap, control subtraction, enrichment trend, and confirmation feasibility, teams can spend substantial time on targets that were never strong enough to justify follow-up.
Sample and Assay Variables That Affect Identification Performance
Before changing platforms, confirm that the starting material and assay conditions fit the biological question.
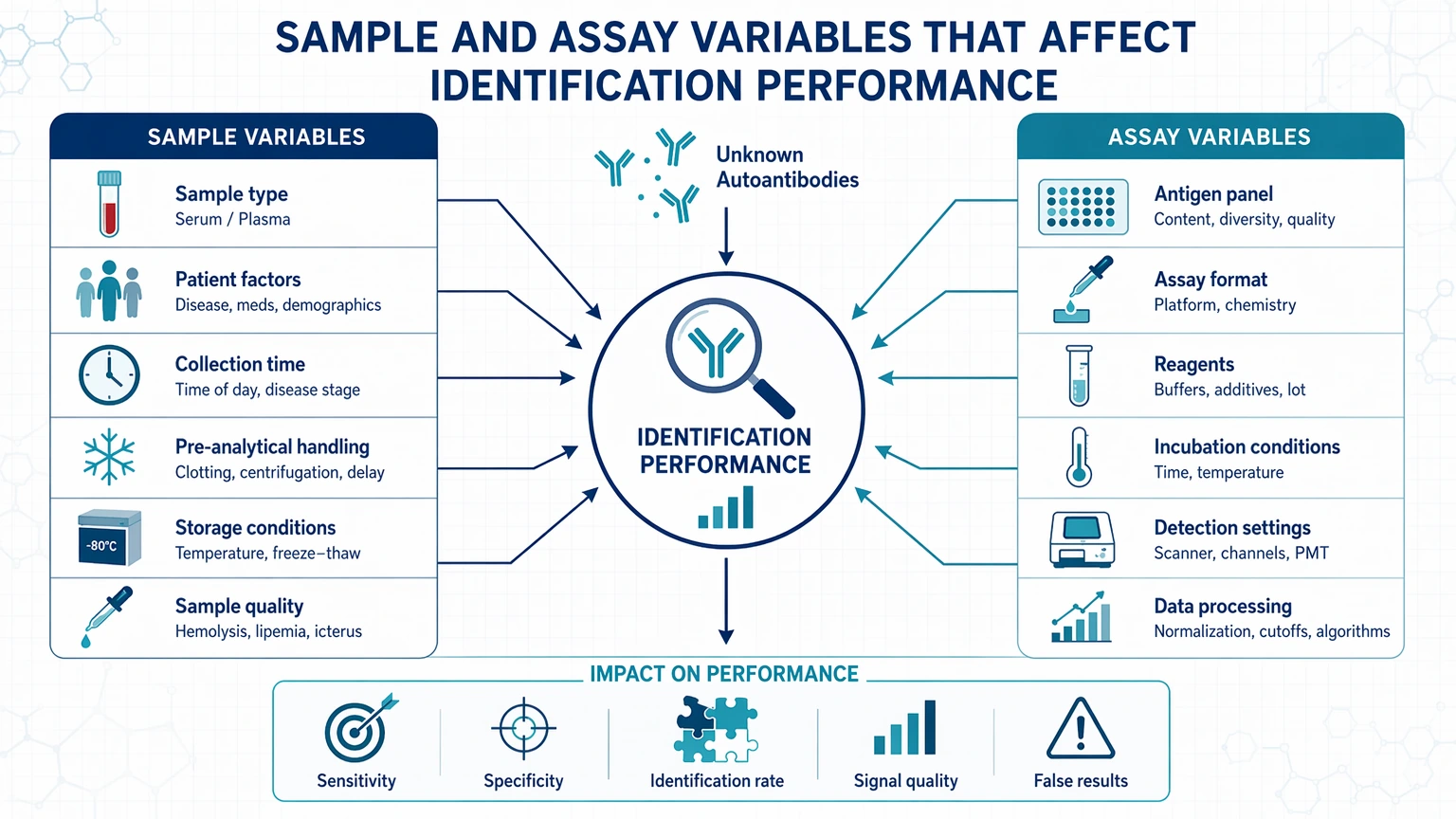
Sample type and handling
Serum, plasma, CSF, and other matrices do not behave the same way. Plasma can introduce anticoagulant-related interference, while serum differs in background protein composition. CSF often contains lower total immunoglobulin input and may require tighter planning for concentration and recovery. Repeated freeze-thaw cycles, prolonged room-temperature handling, and inconsistent aliquoting can reduce functional antibody activity or increase variability between technical repeats.
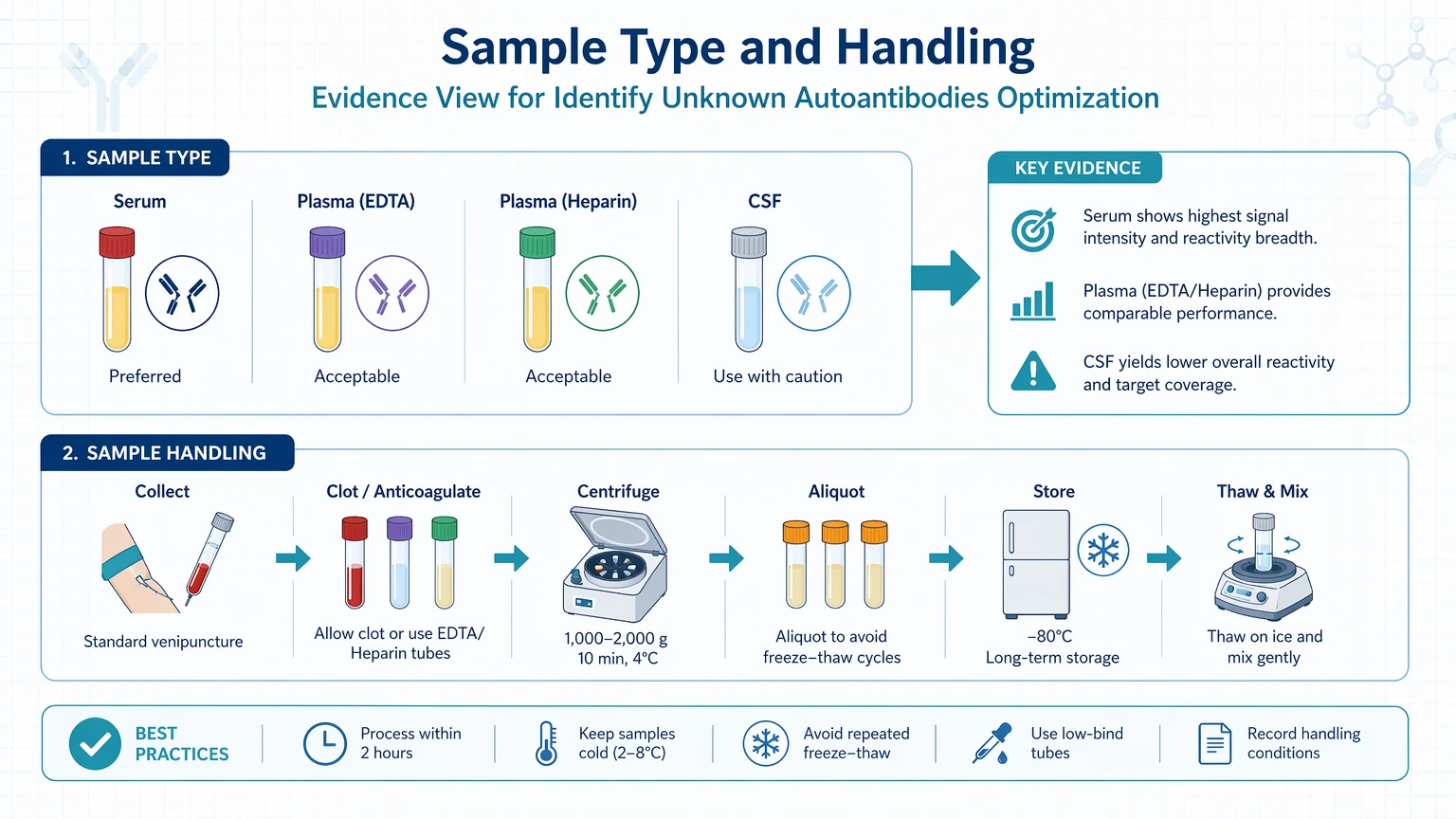
Useful checkpoints include:
Pretreatment and depletion effects
IgG purification, albumin depletion, or fractionation can help, but they can also distort the readout. These steps may improve detectability by reducing matrix complexity, yet they may also bias recovery, remove low-abundance complexes, or alter the native state of interacting molecules. Judge each pretreatment step by its effect on recoverable antibody activity, not just by cleaner chromatograms or lower total protein background.
Epitope presentation
Assay choice should reflect the likely binding mode.
The key question is not which platform is stronger in general. It is which platform preserves the type of epitope presentation most likely to matter in your samples.
How to Optimize Antigen Capture, Screening, and Hit Prioritization
Optimization works best when each stage has a distinct purpose.
1. Improve immune complex enrichment before scaling discovery
If immunoprecipitation is part of the workflow, test enrichment quality before focusing on downstream identification counts. A larger number of proteins is not automatically better. Review whether negative controls pull down similar background, whether wash conditions remove weak contaminants without erasing the expected signal, and whether recovered material is consistent across replicates.
For mass spectrometry readout, useful trend-based indicators include:
2. Reduce non-specific binding at the screening stage
When background dominates, tighten the assay before generating more candidates. Changes may include refined blocking conditions, adjusted sample dilution, stricter wash composition, pre-clearing steps, control of Fc-related interference, or competitor proteins that suppress sticky interactions. Evaluate these changes against negative controls and replicate agreement, not just by a visible drop in total signal.
3. Rank candidates with a predefined filter cascade
Candidate antigen prioritization should happen before interpretation meetings, not during them. A practical filter cascade can include:
This approach avoids promoting candidates because they appear interesting rather than because they survived controlled filtering.
4. Match discovery depth to confirmation capacity
If the team can validate only a small number of targets, the discovery stage should be optimized for confidence ranking rather than maximum list length. A narrower, better-ranked output often supports more efficient target confirmation than a broad screen with unclear selection logic.
When internal data still do not show where confidence is being lost, a project-scoped workflow review can be more useful than another full screening round. At that point, you can submit your requirements and evaluate your project with MtoZ Biolabs in the context of sample type, assay mechanism, and downstream confirmation goals.
How to Confirm Candidate Autoantibody Targets With Orthogonal Methods
Orthogonal validation is where many unknown autoantibody programs either gain traction or reveal that the discovery signal was assay-bound.
A useful confirmation plan changes at least one major variable from the discovery step. That variable might be antigen format, detection mechanism, immobilization context, or enrichment logic. The goal is not perfect agreement. The goal is to test whether the same candidate remains credible under a different experimental assumption.
Examples include:
Interpretation should stay bounded. A candidate that confirms in one orthogonal assay is stronger than a screen-only hit, but it is still a research candidate. A candidate that fails confirmation is not automatically false either; it may indicate that the original assay captured native structure or complex-dependent binding that the follow-up method did not preserve.
Useful confidence checkpoints include:
When to Use External Analytical Support for Complex Unknown Autoantibody Projects
External support becomes useful when the problem is no longer assay execution alone, but workflow architecture. That point often arrives when internal teams face one or more of the following:
A specialized research partner can help separate the problem into practical stages: what to adjust in sample preparation, what to screen in native versus denatured formats, which controls are missing, and which candidate antigens are worth carrying forward. This kind of support is most useful when the team needs project-specific analytical planning rather than generic assay repetition.
If your group is deciding whether to redesign the workflow, narrow the candidate list, or add a different confirmation layer, contact us to discuss the project with the project team and align assay selection with the biology and the sample constraints already in hand.
Key Questions to Ask Before Expanding the Study
Before committing more samples or budget, ask the following:
1. Is the current signal strong enough to justify scale-up, or does it first need cleaner control separation? 2. Does the assay format preserve the suspected epitope type, including conformational antigen or modification-dependent recognition? 3. Are the top candidates reproducible across repeats, or are they driven by one run? 4. Have serum or plasma matrix effects been tested directly rather than assumed away? 5. Does the confirmation method challenge the discovery hit in a meaningful orthogonal way? 6. Can the remaining sample support one more optimization cycle without compromising later validation work?
A “no” on any of these points does not stop the project, but it usually means the next step should be workflow refinement rather than study expansion.
Service Routes for Study Planning
For teams moving from method selection into execution, these service paths connect assay design, validation, and interpretation needs.
Conclusion
The most effective way to improve unknown autoantibody identification is to treat the workflow as a chain of evidence rather than a single experiment. Start by confirming that the sample still contains usable antibody activity. Then choose a discovery format that matches the likely epitope presentation, tighten control of non-specific binding, and filter candidates with predefined criteria before investing in follow-up work. Finally, use orthogonal validation to test whether top hits remain credible under a different assay logic.
This approach fits exploratory and translational research teams that already see some autoantibody-like activity but do not yet have defensible target confirmation. It also has clear limits: no single platform captures every antigen class, conformation, or modification state. When repeated optimization still leaves broad background or weak confirmation, a narrower redesign or staged external review is often more efficient than another full screening round.
FAQ
Why do apparently strong discovery hits disappear during follow-up testing?
The most common reason is a change in antigen context. A discovery assay may present a native complex, a partially folded protein, or a modification-dependent epitope that the follow-up assay does not reproduce. In other cases, the original signal came from non-specific binding that looked convincing only because the controls did not clearly separate background from true enrichment.
How should teams choose between protein microarray, peptide array, and immunoprecipitation?
Choose the first screen based on what kind of binding you expect to preserve. Protein microarrays can be useful when many structured protein candidates need to be surveyed at once. Peptide arrays are more informative when linear epitopes are likely or when a candidate list has already been narrowed. Immunoprecipitation is often the better starting point when the target may require native context or may exist within a protein complex.
What are the clearest signs that serum or plasma matrix effects are distorting the readout?
Matrix interference often shows up as rising background, unstable candidate rank order across replicates, or poor agreement between untreated and pretreated samples. A practical way to test this is to compare performance before and after a controlled pretreatment or dilution change while keeping the same controls and thresholds. If the candidate list shifts substantially without cleaner control separation, the matrix is still shaping the signal.
When does mass spectrometry add the most value to autoantibody identification?
Mass spectrometry adds the most value after enrichment conditions and controls are strong enough to produce interpretable protein lists. It is especially informative in immunoprecipitation-based workflows when the aim is to identify proteins associated with recovered immune complexes. It is less useful when upstream enrichment still carries heavy background, because the downstream list then reflects noise as much as target biology.
How large should the shortlist be before target confirmation begins?
There is no fixed number, but the shortlist should be small enough that each retained candidate has a clear technical rationale. A workable confirmation set usually includes targets supported by repeatability, control separation, assay-fit logic, and a biologically plausible reason for follow-up. A shorter, higher-confidence list often leads to better project decisions than a long panel of marginal hits.
How to order?

