





Glycosylation Quantitative Proteomic Analysis Service
Protein glycosylation is a common type of post-translational modification (PTM) involving the covalent bonding of oligosaccharide chains to polypeptide chains. The synthesis pathway of mammalian glycans consists of 10 monosaccharide units: fucose (Fuc), galactose (Gal), glucose (Glc), N-acetylgalactosamine (GalNAc), N-acetylglucosamine (GlcNAc), glucuronic acid (GLCA), iduronic acid (IdoA), mannose (Man), sialic acid (SA), and xylose (Xyl). It is estimated that mammals require about 700 proteins and over 7,000 different structures to produce complete glycan diversity. Protein glycosylation in humans includes N-linked glycosylation, O-linked glycosylation, and C-glycosylation, among which N-linked and O-linked glycosylation are the most common types. N-linked glycosylation occurs on asparagine with the characteristic sequence Asn-X-Ser/Thr (and scarcely Cys), where X is any amino acid except proline. N-linked glycans usually have a core pentasaccharide moiety and are classified by the type and position of monosaccharide residues added to the core pentasaccharide. O-linked glycosylation is located on serine or threonine residues, initiated by adding GalNAc, Man, Fuc, Glc, or GlcNAc.
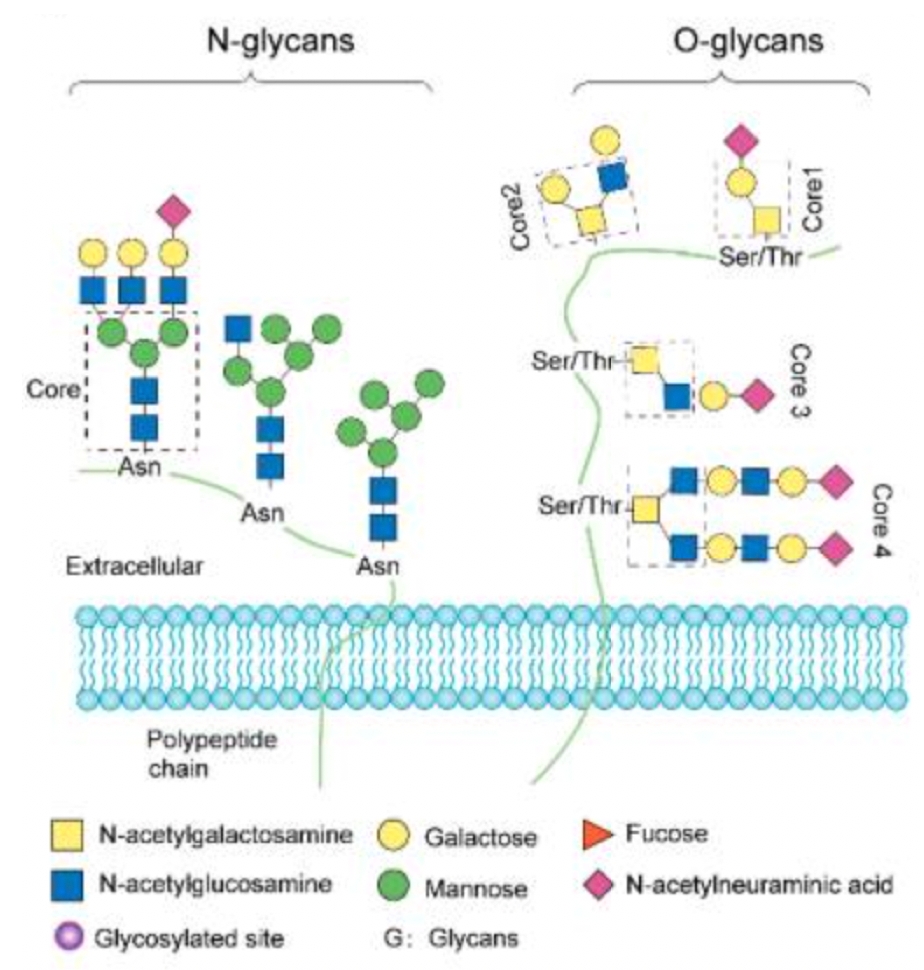 Figure 1. Common Types of Glycosylation Modifications [3]
Figure 1. Common Types of Glycosylation Modifications [3]
Abnormal glycosylation in glycoproteins may be associated with the occurrence and development of certain diseases. Abnormal glycosylation patterns, such as increased glycan size and extra branching of glycan chains, accompanied by over-sialylation and -fucosylation, are closely related to disease progression. As the progression of some diseases may involve abnormal glycosylation in glycoproteins, a tool that can quantitatively analyze proteins with specific glycan structures can monitor differences between individual cases in the abundance of abnormal glycoforms of disease-related target proteins. Thus, effectively identifying these glycoproteins and quantifying the levels of glycoproteins between healthy and malignant individuals help understand the pathogenic mechanisms of malignant cells and ultimately determine specific disease biomarkers.
Figure 2. Applications of Glycosylation Quantitative Proteomics in Disease Research [4]
Glycoproteins can be analyzed through the following steps: (1) protein extraction from organisms; (2) digestion of proteins; (3) enrichment of glycopeptides; (4) fractionation of glycopeptides by high pH RPLC; (5) detection of glycopeptides by liquid chromatography-tandem mass spectrometry (LC-MS/MS); (6) data analysis. If quantitative analysis is required, label-based or label-free methods can be used.
1. Protein Extraction and Digestion
Lysis buffer may vary depending on the protein application. For example, buffers containing Tris are mainly used for extracting proteins from cells or tissues for SDS-PAGE, glycoprotein staining, glycoproteomics, and proteomics. When glycoproteins are immobilized on aldehyde resin for glycan enrichment, it is preferable to choose buffers without amines, as aldehyde groups react with amines (such as Tris, ammonia, urea), reducing the efficiency of protein immobilization.
Bottom-up glycoproteomics requires the use of proteases to digest glycoproteins into shorter peptides and glycopeptides. The choice of protease depends on the targeted protein sequence and its structure to be discovered. Pepsin can cleave peptide Phe(F)/Leu(L) residues at pH=1.3, F/L/Trp(W)/Tyr(Y) at pH>2. Caspases 1 to 7 can cleave the C-terminal of Asp(D), but have different specificities for protein substrates. Caspase 8 preferentially cuts P1 of peptides composed of IETD|X or LETD|X. Caspase 9 cuts LEHD|X, and caspase 10 cuts IEAD|X. Chymotrypsin has high specificity for Trp, Tyr, and Phe at the P1 position, but lower specificity for Leu, Met, and His. When His, Lys, or Arg is at the P2 position, prolyl endopeptidase cleaves the C-terminal of Pro at the P1 position. Trypsin is the most commonly used protease, which can cleave the C-terminal of Lys or Arg at the P1 position. Hydroxylamine can cleave the C-terminal of Asn, and thrombin can digest Arg at the P2 position of Gly. Other types of enzymes cleave at the P1 position. Asp-N endopeptidase and formic acid digest Asp. Arg-C proteinase, and clostripain cleave Arg. BNPS-Skatole, and iodobenzoic acid digest Trp (W), and protease K can cleave multiple amino acids at the P1 position.
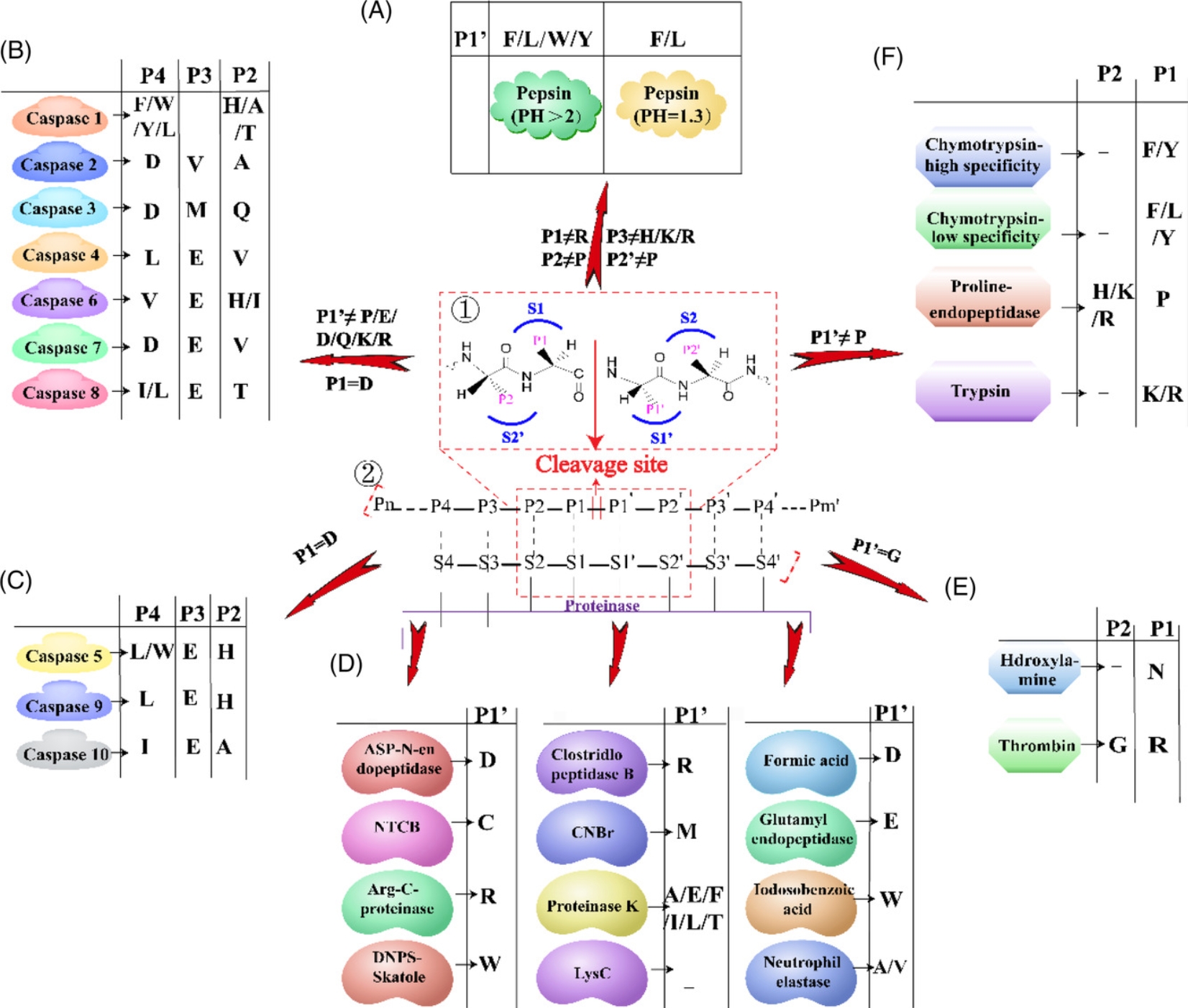 Figure 3. Cleavage Sites of Different Proteases [5]
Figure 3. Cleavage Sites of Different Proteases [5]
2. Glycopeptide Enrichment
Several strategies for enriching glycopeptides have been developed, including hydrophilic interaction liquid chromatography (HILIC), lectin affinity chromatography (LAC), chemical coupling, boronate affinity, and metabolic labeling.
(1) Hydrazide-Capturing Technique
The hydrazide-capturing technique is a chemical reaction-based method that binds hydrazide groups fixed on beads to dialdehyde groups from glycan moiety, enriching glycoproteins or glycopeptides by selectively targeting cis-diols on monosaccharides in glycan moieties. In the hydrazide chemistry, cis-diols on glycan are oxidized into aldehydes by sodium periodate. Then it is covalently coupled to hydrazide fixed on solid beads. The bound glycoproteins are generally digested in situ by trypsin, and washed to remove non-glycosylated or unbound peptides. The covalently coupled glycopeptides are then released by PNGase F and analyzed by MS for deglycosylated peptides. During the PNGase F-mediated enzymatic process, the asparagine residue in the NX(S/T) consensus sequence captured by N-glycosidic bond hydrazide is converted to an aspartic acid with COOH functionality at side chain.
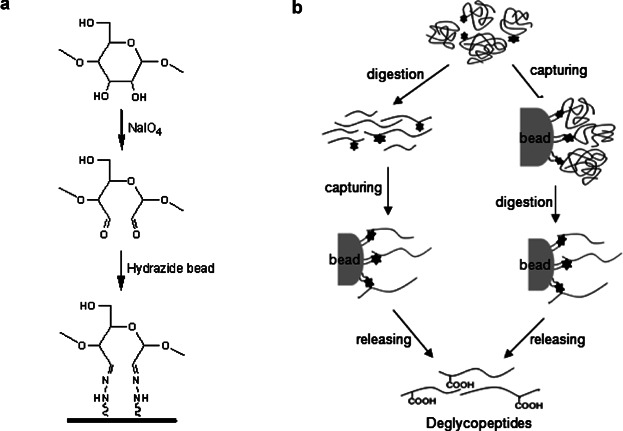 Figure 4. Hydrazide Capture Method for Glycoproteome Enrichment [6]
Figure 4. Hydrazide Capture Method for Glycoproteome Enrichment [6]
The hydrazide chemistry-based hydrazide capture method can be used to capture glycosylated molecules from highly complex samples and characterize the captured molecules with high-throughput analytical performance. Nevertheless, because this hydrazide-based method utilizes dialdehyde groups produced by oxidative cleavage of glycans from glycoproteins or glycopeptides to capture the glycans, structural information on the complete glycan structure of glycoproteins or glycopeptides is inevitably lost. Moreover, these chemical reaction-based method are primarily selective for cis-diols on monosaccharides composed of glycans, thus, using only this hydrazide capture method cannot specifically capture protein glycoforms present in abnormal biological processes.
(2) Hydrophilic Interaction Liquid Chromatography (HILIC)
HILIC is considered an effective method for enriching and isolating glycopeptides and glycans in glycoproteins. HILIC features a hydrophilic stationary phase (based on functionality of cation exchange, anionic exchange, copolymer interaction, and sepharose, etc.) and a relatively hydrophobic organic mobile phase. Due to the influence of hydrophilic carbohydrate molecules, glycopeptides most are strongly retained on the HILIC chromatography column and separated from the non-glycosylated peptides left in the digest. Thus, each glycan or glycopeptide can be analyzed through chromatographic analysis and MS detection. Combining HILIC with other techniques (such as glycoprotein hydrazide capture and lectin-specific separation) is attractive for subsequent MS-based analysis of complex glycoproteomes in the field of glycoproteomics.
(3) Lectin-Specific Capturing Techniques
Various lectins have been widely used to capture glycoforms of proteins, which have specific binding affinity with the lectins used. Its most notable feature is the ability to enrich unique protein glycoforms that have specific glycan structures action for the lectins used. Lectins mainly bind with five specific carbohydrates, including mannose, galactose or GalNAc, GlcNAc, fucose, and sialic acid.
Because one major feature of lectin capture is its ability to bind with glycoproteins based on the structural features of the glycoprotein, a single lectin capture method can fractionate each specific protein glycoform from complex proteome samples separately for each lectin used, rather than separating the total glycoforms from the proteome samples. Another feature of lectin capture is the elimination of the negative masking effect of protein glycoforms that are inactive for the lectins used. In addition to using a single lectin or a pre-mixed multi-lectin column, a multi-lectin channel monitoring method can also be used, which is based on multiple parallel lectin captures, separately fractionating protein glycoforms active for each lectin used.
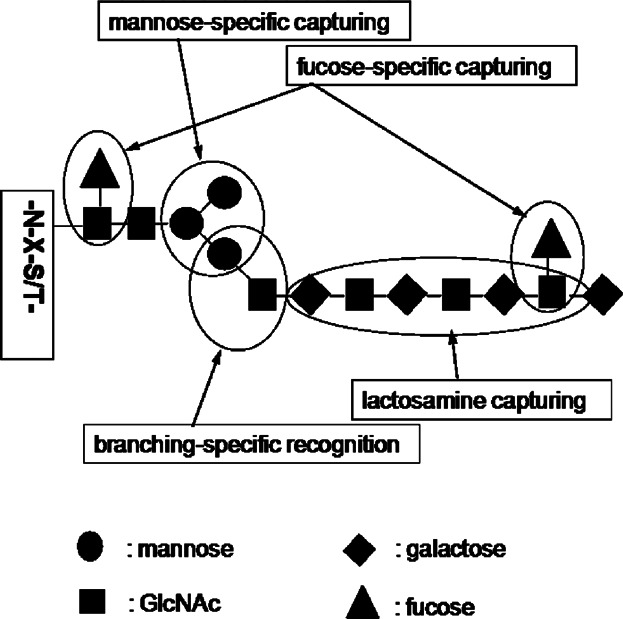 Figure 5. Lectin Specificity Capture Technique for Glycoproteome Enrichment [6]
Figure 5. Lectin Specificity Capture Technique for Glycoproteome Enrichment [6]
(4) Affinity Separation Using Porous Graphite Carbon
Porous graphite carbon is attractive for capturing and separating hydrophilic glycans released from glycoproteins but not retained by reverse-phase chromatography. The separated glycans can be analyzed by mass spectrometry as free glycans or as derivatives labeled with various tagging techniques (such as permethylation, peracylation) to achieve high-sensitivity detection or improved chromatographic separation of the glycans to be analyzed. Graphite carbon can also be used to analyze complex glycopeptide mixtures. Glycopeptides produced by enzymatic hydrolysis with various endoproteases (such as trypsin) from the total proteome or fractionated glycoproteome can be targeted, rather than glycan moieties. Before tandem mass spectrometry-based analysis, the glycopeptides are also routinely separated at the peptide level by porous graphite carbon and chromatography. However, this glycan-targeted approach has a drawback in that the information on the specific glycan components of glycoproteins or glycosylation sites inevitably disappears during the preparation of glycans by enzymatic or chemical deglycosylation reactions. Therefore, this method can serve as a supplementary method to the glycan-targeted approach to analyze the deglycosylated protein part of glycoproteins by deglycosylation, aiding in the identification of glycoproteins and glycosite complexes.
(5) Boronic Acid Affinity Separation Technique
Boronic acid can enrich glycoproteins or glycopeptides by selectively capturing glycan moieties. It binds cis-diols in monosaccharides on glycan moieties selectively through forming boronate heterocyclic diesters. The glycoproteins or glycopeptides captured by boronic acid are released from it by exchanging with monosaccharides in a high-concentration elution buffer after purification with washing buffer. Generally, the boronic acid fixed on magnetic beads captures glycoproteins and glycopeptides by forming boronate diesters with vicinal diols on glycan moieties. Moreover, boronic acid-modified gold nanoparticles or gold-coated silicon water are used for enriching glycopeptides and subsequent matrix-assisted laser desorption/ionization (MALDI) MS. There are also studies developing boronate affifinity monolithic columns for selective enrichment of glycopeptides and glycoproteins, and their capturing performance is evaluated by capillary liquid chromatography (LC). These boronic acid capture methods are suitable for capturing glycosylated molecules from highly complex samples with complete glycan structures and characterizing the captured glycans with high-throughput analytical performance. However, the boronic acid capture method mainly has selectivity only for cis-diols on monosaccharides composed of glycans but is not optimistic about selectively capturing glycosylated forms of proteins with specific glycan structures.
(6) Metabolic Labeling Glycosylation Analysis
Metabolic labeling is a method of incorporating labels into endogenous synthesis and modification mechanisms in cells. In proteomics, cells use these analogs to synthesize and modify proteins, targeting cellular proteins, thus selectively enriching glycoproteins. Analogues such as N-azidoacetyl galactosamine-tetraacylated (Ac4GalNAz) have been incorporated into cells, forming GalNAz-modified glycoproteins, which can be specifically separated using a Staudinger ligation (azide-phosphine connection). The azide-labeled glycoprotein complexes can conjugate with alkyne-containing probes for comprehensive identification of complete N-glycopeptides and O-glycopeptides. Similar to SILAC (stable isotope labeling by amino acids in cell culture) labeling, this strategy is not applicable to human tissues or any clinical samples.
3. Label-Based Quantitative Glycoproteomics Analysis Workflow
With the development of proteomics, quantitative glycoproteomics methods commonly use stable isotopes such as 13C, 15N, 18O, and 2H to label glycopeptides. This labeling leads to mass shifts of the glycopeptide precursors or fragments, allowing glycopeptides from different samples to be labeled differently and mixed before measurement, yet still distinguishable by MS. Stable isotopes can be incorporated into glycopeptides through chemical, metabolic, or enzymatic means. These strategies can further be classified into isotopic or isobaric labeling, where the relative quantification of labeled glycopeptides is achieved at the MS1 or MSn level.
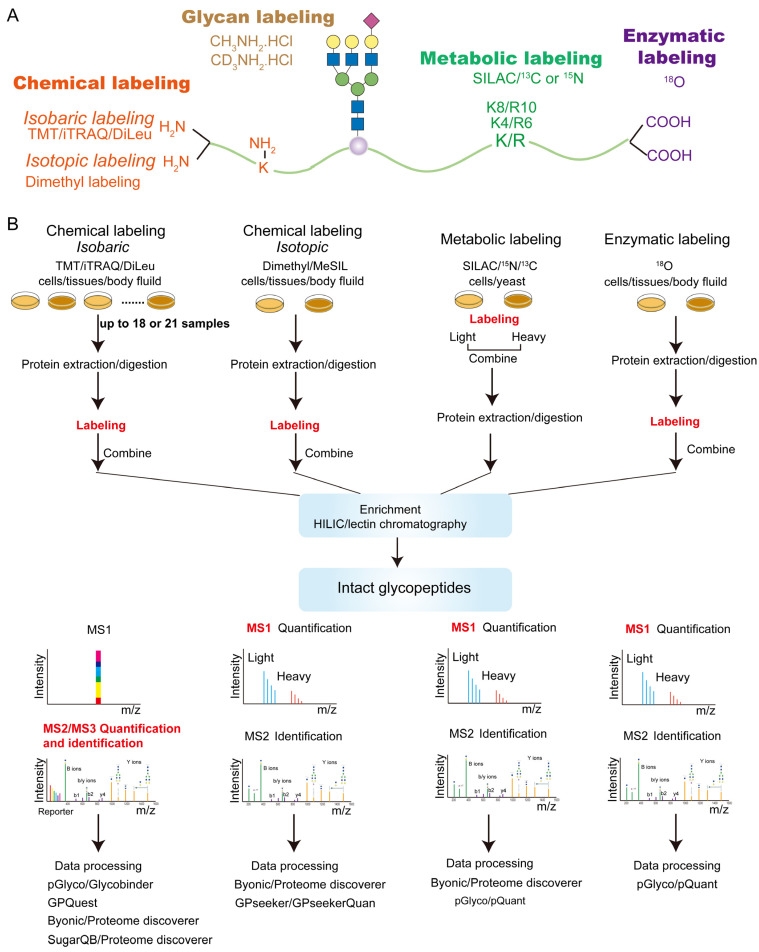 Figure 6. Label-Based Quantitative Glycoproteomics Strategy [3]
Figure 6. Label-Based Quantitative Glycoproteomics Strategy [3]
4. Label-Free Glycoproteomics Quantitative Analysis Workflow
Label-free quantification (LFQ) methods aim to quantify glycopeptides without using stable isotope labeling. Samples to be compared are prepared separately, including protein extraction, digestion, glycopeptide enrichment, and parallel LC-MS/MS measurement. The extracted ion currents (XIC) during different runs or identified glycopeptide spectral counts are compared. Depending on the mass spectrometry acquisition method, LFQ can be divided into data-dependent acquisition (DDA) and data-independent acquisition (DIA) quantitative analysis. In DDA mode, the most abundant precursor ions detected in MS1 survey scans are selected and sequentially isolated within a narrow mass-to-charge (m/z) window for MS2 fragmentation. The number of precursor ions selected per acquisition cycle depends on the survey scan and predefined settings. However, in DIA mode, the precursor isolation range, MS2 event number, and precursor m/z coverage are predefined. Each acquisition cycle repeats the same scanning events regardless of the collected data.
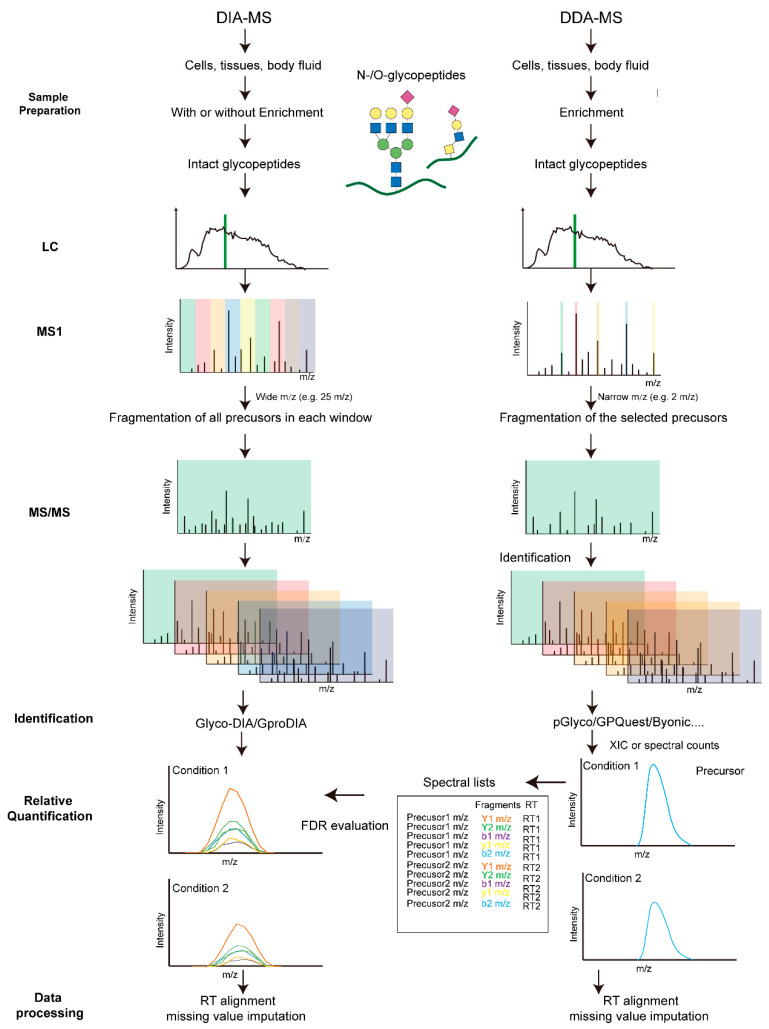
Figure 7. LFQ Glycoproteomics Strategy [3]
Analysis Workflow
1. According to the Experimental Requirements to Determine the Experimental Process
2. Mass Spectrometry Sample Preparation
3. High-resolution Mass Spectrometry Acquisition Data
4. Data Retrieval and Analysis
Service Advantages
1. Identification/Quantification/Modification Identification of Multi-Type Sample Source Proteins
2. High-confidence, High-precision Mass Spectrometry Detection
3. Comprehensive Bioinformatics Analysis
Sample Results
1. Comprehensive Analysis of Glycoproteins Secreted by Cultured Cells without Serum Starvation[7]
Glycoproteins secreted by cells play a crucial role in regulating extracellular activities. Secreted glycoproteins often reflect the state of cells, thus glycoproteins extracted from easily accessible body fluids can serve as excellent biomarkers for disease detection. In the fields of biology and biomedical research, cultured cells have been widely used as models to thoroughly analyze these secreted glycoproteins, gaining insights into cellular activities and glycoprotein functions. However, compared to the high-abundance serum proteins necessary for cell growth and proliferation, secreted glycoproteins are of lower abundance, making the comprehensive identification and quantification of secreted glycoproteins a daunting task. Some studies use serum-free culture media to analyze secreted proteins, but research shows that even short-term serum starvation can change protein secretion. To overcome these issues, a method has been developed that combines selective enrichment and enhancement techniques for secreted glycoproteins from cultured cells, identifying and quantifying their N-glycosylation sites comprehensively. Results indicate that enhancing sample selection and enhancement relative to the sample ratio is crucial for improving the coverage of secreted glycoproteins. This method has been applied to global quantitative analysis of secreted glycoproteins in response to lipopolysaccharide (LPS) in THP-1 monocytes and macrophages, as well as in Hep G2 cells treated with TGF-β and subjected to serum starvation. Different secreted glycoproteins found in these model systems indicate cellular responses to immune activation or epithelial-to-mesenchymal transition. Thanks to the selective enrichment and signal enhancement of low-abundance secreted glycoproteins, this method can be widely applied to the study of serum-starved secreted glycoproteins, thus better understanding protein secretion and cellular activity.
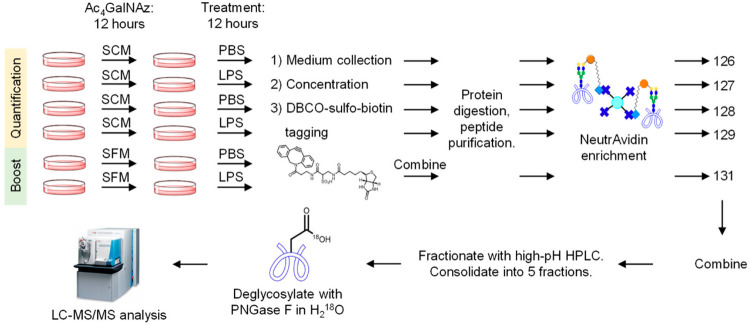
Figure 8. Quantitative Analysis of Cell-Secreted Glycoproteins
2. A Simplified Workflow for Multiplex Quantitative Site-Specific N-glycoproteomics[8]
Regulation of protein N-glycosylation is crucial in human cells. However, large-scale, accurate, and specific site quantification of glycosylation remains technically challenging. A study introduced SugarQuant, a mass spectrometry-based integrated pipeline comprising sample preparation based on protein aggregation capture (PAC), multi-notch MS3 acquisition (Glyco-SPS-MS3), and data processing tools (GlycoBinder), which reliably identified and quantified complete glycopeptides in complex biological samples. PAC significantly reduced sample processing time without affecting sensitivity. Glyco-SPS-MS3 combined high-resolution MS2 and MS3 scans, thus enhancing the isobaric mass tag reporting signal, improving detection of N-glycopeptide fragments, and reducing interference in multiplex quantification. GlycoBinder simplified the data processing of Glyco-SPS-MS3 data, followed by a two-step database search, which increased the identification rate of glycopeptides by 22% compared to traditional methods. SugarQuant has been applied to identify and quantify over 5,000 unique glycoforms in Burkitt's lymphoma cells and to determine specific site glycosylation changes following inhibition of fucosylation at high confidence.
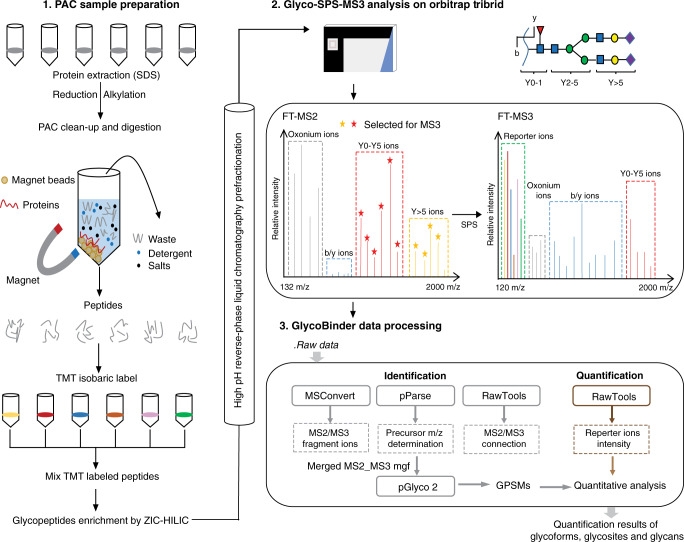
Figure 9. SugarQuant Analysis Process
3. GproDIA for Comprehensive Statistical Control in Data-Independent Acquisition Glycoproteomics[9]
Large-scale analysis of complete glycopeptides is essential in glycoproteomics but poses challenges. DIA is an emerging technique in proteomics with deep proteome coverage and accurate quantitation capabilities, but it is still in its early development stage in glycoproteomics. A study introduced GproDIA, a framework for characterizing complete glycopeptides from DIA data, using a two-dimensional false discovery rate method and glycoform inference algorithms for comprehensive statistical control, enabling accurate identification of complete glycopeptides using wide isolation windows. Further, a semi-empirical spectrum prediction strategy is employed to expand the coverage of the spectral glycopeptide library. The study benchmarked N-glycopeptide analysis methods on yeast and human serum samples' DIA data, showing that GproDIA's DIA outperforms in identification capability and data integrity, as well as in accuracy and precision of quantitation, compared to the data-dependent acquisition-based methods for glycoproteomics.
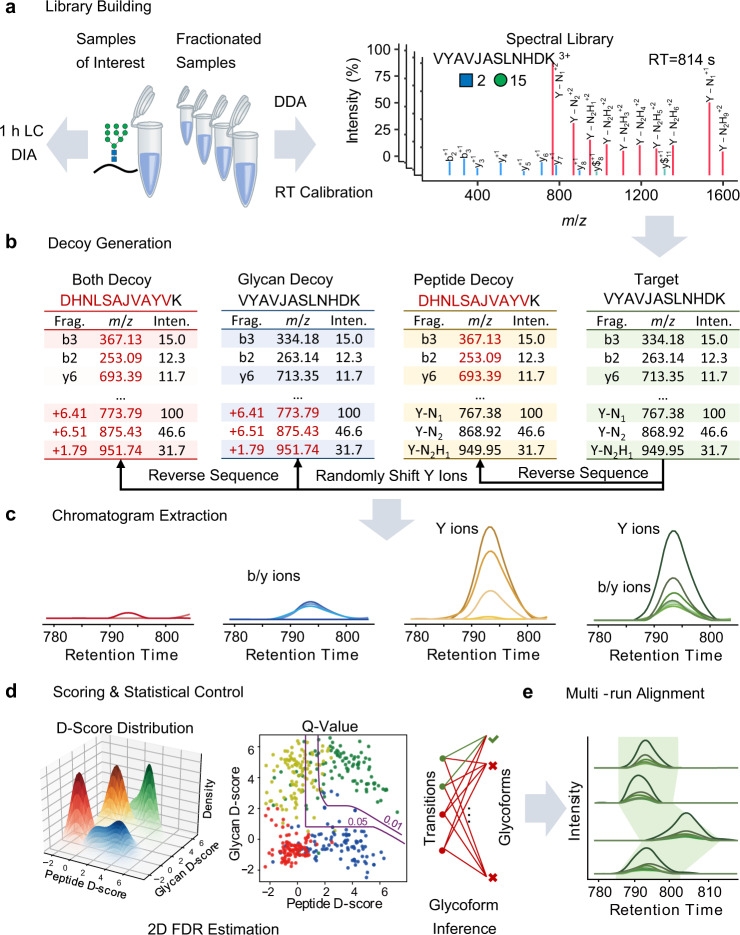
Figure 10. GproDIA Analysis Process
4. Quantitative Study of Human Cell Surface N-glycoprotein Dynamics[10]
Surface glycoproteins regulate almost every extracellular event, actively adapting cells to continuously changing extracellular environments. These glycoproteins contain a wealth of information about cell development and disease states, having significant biomedical relevance. Systematic study of surface glycoproteins helps better understand the functions of surface proteins, cell activities, and the molecular mechanisms of diseases. However, specific and global analysis of surface glycoproteins is a highly challenging task. A study designed a method that integrated pulse-chase labeling, selective enrichment of surface glycoproteins, and multiplexed proteomics to systematically analyze the dynamics of surface glycoproteins and measure their half-lives. The study results clearly demonstrated that surface glycoproteins with catalytic activity were more stable than those with binding and receptor activities. Glycosylation sites located outside any structural domain had a significantly longer median half-life than those within domains, strongly suggesting that glycans within domains regulate protein interactions with other molecules, while those outside of domains primarily served to protect the protein from degradation. This method can be broadly applied in biological and biomedical research.
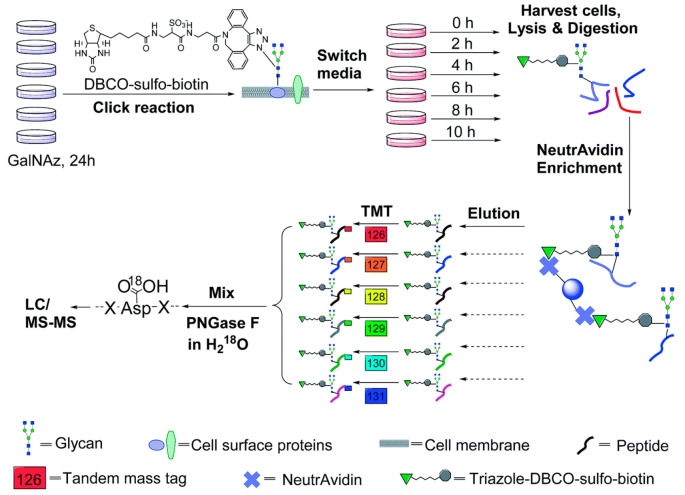
Figure 11. Quantitative Study of Cell Surface N-Glycoprotein Dynamics
Sample Submission Requirements
1. Protein Purity >90%.
2. Try to Avoid Impurity Contamination
Services at MtoZ Biolabs
1. Experimental Procedure
2. Relevant Mass Spectrometry Parameters
3. Raw Data
4. Data Analysis Reports
Applications
1. Mining of Disease Biomarkers
Personalized medicine has become a widely accepted trend in medicine, which is used for the effective and safe treatment of various diseases. It encompasses tailor-made medical care for each individual based on their various characteristics. Tumor-related glycosylation is a major theme in glycoproteomics, involving the identification of abnormal glycoproteins, the structural analysis of protein glycans, and their impact on diseases. Cancer-related glycosylation more precisely reflects the cancer state, and special glycan binding probes (usually lectins) can be paired with glycosylation antibodies to achieve quantitative and qualitative measurements of glycosylation.
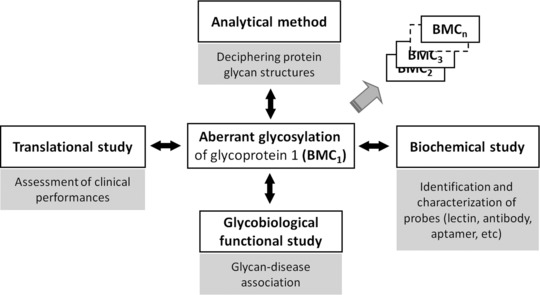
Figure 12. Mining of Glycoprotein-Related Disease Biomarkers [11]
2. Research on Disease Mechanisms
Glycosylation process in cells is highly sensitive to physiological states, and glycans are a common disease biomarker. Extensive use of lectins, antibodies, and direct structural analysis of glycans has documented human diseases, especially the various changes in glycosylation in cancer. However, insights into how these changes in glycosylation occur, what the functional consequences, and the nature of specific molecular mechanisms is still limited. Exploring the regulation of glycosylation through quantitative proteomics data opens new pathways for understanding the structure-function relationships of protein glycosylation.
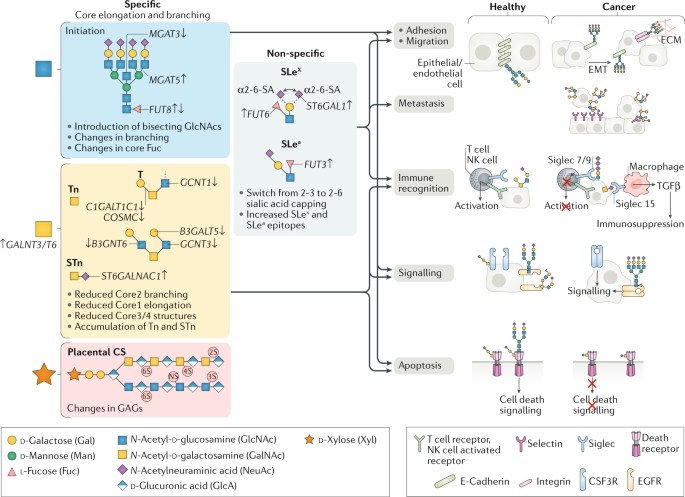
Figure 13. Three Main Glycosylation Pathways Undergoing Characteristic Changes in Cancer [12]
FAQ
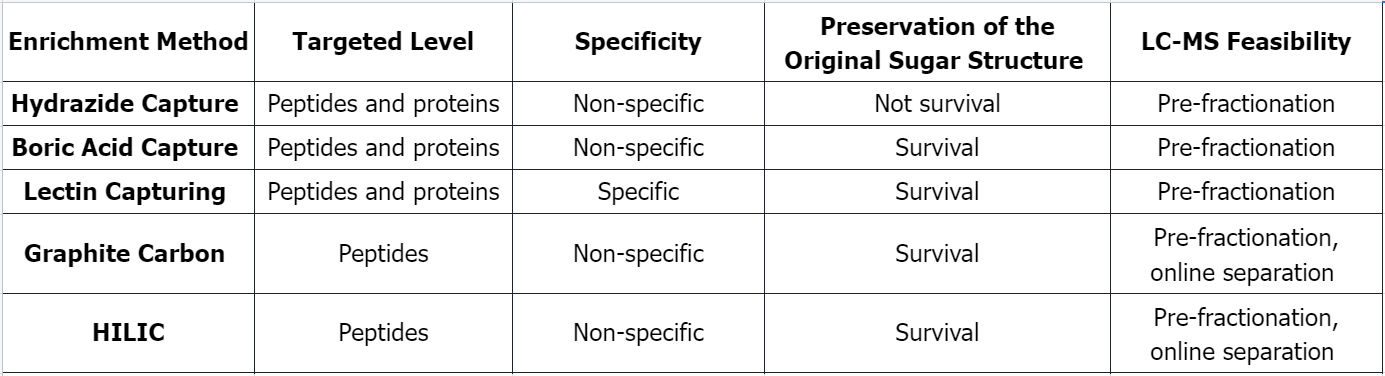
Q1: Comparison of Several Common Glycosylated Peptide Enrichment Methods
References
[1] CoxJ, Mann M. Quantitative, high-resolution proteomics for data-driven systems biology. Annu Rev Biochem. 2011;80:273-99. doi: 10.1146/annurev-biochem-061308-093216. PMID: 21548781.
[2] Ahire D, Kruger L, Sharma S, Mettu VS, Basit A, Prasad B. Quantitative Proteomics in Translational Absorption, Distribution, Metabolism, and Excretion and Precision Medicine. Pharmacol Rev. 2022 Jul;74(3):769-796. doi: 10.1124/pharmrev.121.000449. PMID: 35738681; PMCID: PMC9553121.
[3] Fang P, Ji Y, Oellerich T, Urlaub H, Pan KT. Strategies for Proteome-Wide Quantification of Glycosylation Macro- and Micro-Heterogeneity. Int J Mol Sci. 2022 Jan 30;23(3):1609. doi: 10.3390/ijms23031609. PMID: 35163546; PMCID: PMC8835892.
[4] Xu S, Xu X, Wu R. Deciphering the Properties and Functions of Glycoproteins Using Quantitative Proteomics. J Proteome Res. 2023 Jun 2;22(6):1571-1588. doi: 10.1021/acs.jproteome.3c00015. Epub 2023 Apr 3. PMID: 37010087; PMCID: PMC10243117.
[5] Li J, Zhang J, Xu M, Yang Z, Yue S, Zhou W, Gui C, Zhang H, Li S, Wang PG, Yang S. Advances in glycopeptide enrichment methods for the analysis of protein glycosylation over the past decade. J Sep Sci. 2022 Aug;45(16):3169-3186. doi: 10.1002/jssc.202200292. Epub 2022 Jul 18. PMID: 35816156.
[6] Ahn YH, Kim JY, Yoo JS. Quantitative mass spectrometric analysis of glycoproteins combined with enrichment methods. Mass Spectrom Rev. 2015 Mar-Apr;34(2):148-65. doi: 10.1002/mas.21428. Epub 2014 Jun 2. PMID: 24889823; PMCID: PMC4340049.
[7] Suttapitugsakul S, Tong M, Sun F, Wu R. Enhancing Comprehensive Analysis of Secreted Glycoproteins from Cultured Cells without Serum Starvation. Anal Chem. 2021 Feb 2;93(4):2694-2705. doi: 10.1021/acs.analchem.0c05126. Epub 2021 Jan 4. PMID: 33397101; PMCID: PMC8034805.
[8] Fang P, Ji Y, Silbern I, Doebele C, Ninov M, Lenz C, Oellerich T, Pan KT, Urlaub H. A streamlined pipeline for multiplexed quantitative site-specific N-glycoproteomics. Nat Commun. 2020 Oct 19;11(1):5268. doi: 10.1038/s41467-020-19052-w. PMID: 33077710; PMCID: PMC7572468.
[9] Yang Y, Yan G, Kong S, Wu M, Yang P, Cao W, Qiao L. GproDIA enables data-independent acquisition glycoproteomics with comprehensive statistical control. Nat Commun. 2021 Oct 18;12(1):6073. doi: 10.1038/s41467-021-26246-3. PMID: 34663801; PMCID: PMC8523693.
[10] Xiao H, Wu R. Quantitative investigation of human cell surface N-glycoprotein dynamics. Chem Sci. 2017 Jan 1;8(1):268-277. doi: 10.1039/c6sc01814a. Epub 2016 Aug 15. PMID: 28616130; PMCID: PMC5458730.
[11] Kang JG, Ko JH, Kim YS. Application of cancer-associated glycoforms and glycan-binding probes to an in vitro diagnostic multivariate index assay for precise diagnoses of cancer. Proteomics. 2016 Dec;16(24):3062-3072. doi: 10.1002/pmic.201500553. Epub 2016 May 11. PMID: 27005968; PMCID: PMC5217075.
[12] Schjoldager KT, Narimatsu Y, Joshi HJ, Clausen H. Global view of human protein glycosylation pathways and functions. Nat Rev Mol Cell Biol. 2020 Dec;21(12):729-749. doi: 10.1038/s41580-020-00294-x. Epub 2020 Oct 21. PMID: 33087899.
MtoZ Biolabs, an integrated chromatography and mass spectrometry (MS) services provider.
Related Services
How to order?

