





Phosphorylation Quantitative Proteomic Analysis Service
Phosphorylation is the most common type of post-translational modification (PTM) that proteins undergo throughout their lifespan. The reversibility and transient nature of phosphorylated protein allow signaling pathways to perform various cellular functions. From bacteria to humans, phosphorylation alters protein functions by changing protein stability, cellular localization, substrate affinity, complex formation, and activity, thereby enabling fundamental events such as the cell cycle and growth to occur at precise times and locations. Activities such as DNA transcription, cell cycle regulation, and energy metabolism, as well as cellular processes such as neuronal migration, immune system recognition, and apoptosis, are initiated or controlled by protein phosphorylation events.
The addition of phosphate groups to substrate proteins is accomplished by protein kinases, which account for 1.5-2.5% of the entire genome in nearly all organisms. The human kinome consists of 518 protein kinases, while drosophila has 228, and yeast 130. It is estimated that the human phosphoproteome contains between 100,000–200,000 phosphorylation sites. Most unique protein kinase families found in mammals play significant roles in the expansion of immune responses or the development of the nervous system. Mutations or dysregulation of protein kinases play a central role in the etiology of many human diseases. In recent years, mass spectrometry-based proteomics analyses have led to the identification of thousands of phosphorylation sites across the proteomes of various species. In future large-scale experiments, combining data-driven modeling strategies based on quantitative data, targeted kinase-substrate screening, and validation in biochemical and genetic experiments will be necessary to specifically discover phosphorylation sites that play direct roles in the regulation of signaling pathways.
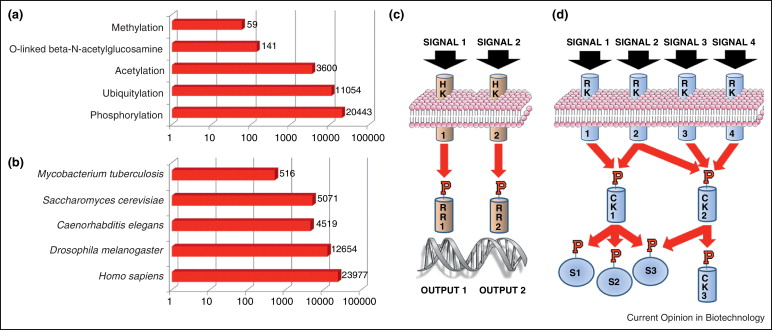
Figure 1. Current Status and Function of Phosphorylation Research [3]
The analysis of protein phosphorylation through bottom-up mass spectrometry-based proteomics primarily involves: (1) Sample preparation; (2) Data collection; (3) Data analysis.
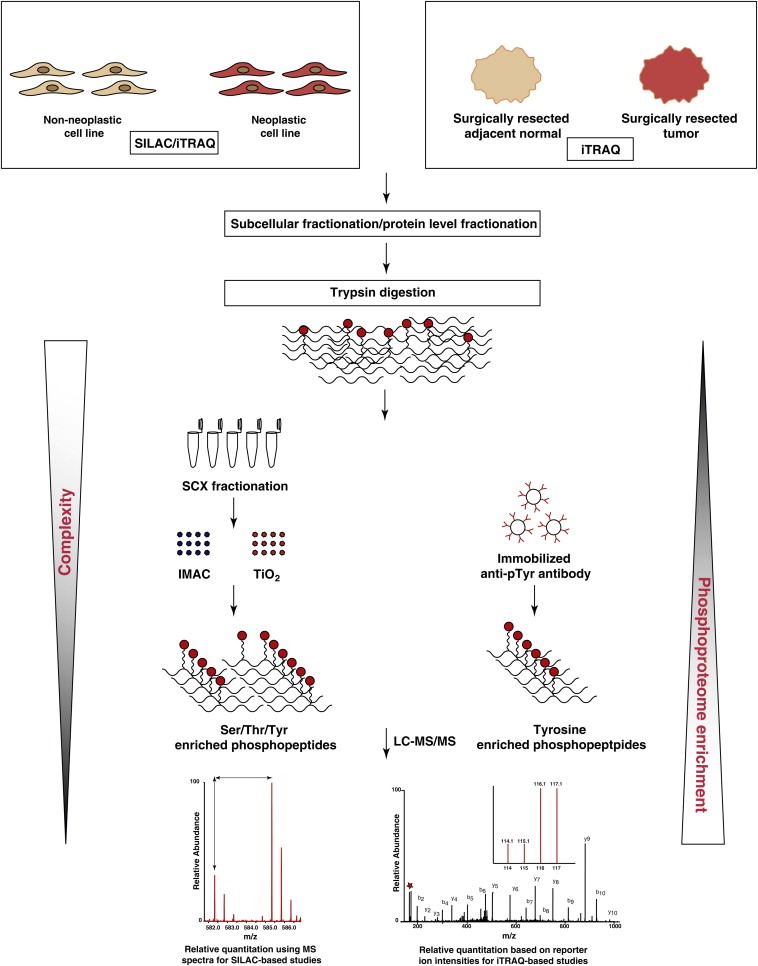
Figure 2. Basic Flow of Quantitative Phosphoproteomics [4]
1. Sample Preparation
Sample preparation is considered the most important aspect of any proteomics experiment. Although advanced instrumentation and data analysis technologies are employed, poor sample quality can greatly impact all followed processes. Since the phosphoryl groups on proteins are chemically stable at acidic and physiological pH, dephosphorylation of phosphoproteins can only occur through the action of phosphatases. Therefore, all sample preparation should be performed on ice to minimize phosphatase activity, and should include a mixture of phosphatase inhibitors to prevent dephosphorylation. Depending on the biological question of interest, specific cellular substructures or organelles can also be isolated and then analyzed to assess their phosphorylation spectrum under various conditions. Due to the low stoichiometry of protein phosphorylation, specific enrichment techniques need to be developed, which are usually applied at the peptide level (after tryptic digestion) to enrich phosphopeptides.
(1) Immobilized Metal Affinity Chromatography (IMAC)
This method relies on the high affinity of phosphopeptides for trivalent metal ions (including Fe3+, Ga3+, and Al3+). The metal ions are fixed in the resin, which contains chelating agents such as nitrilotriacetic acid (NTA) and iminodiacetic acid (IDA). Because acidic peptides can also bind to IMAC materials, it is recommended to esterify the side chains of aspartic acid and glutamic acid as well as the carboxyl group at the C-terminus to minimize non-specific binding of acidic peptides. To enhance the specificity and robustness of phospho-enrichment, several improvements have been made to the IMAC method. Elements like nickel are linked to NTA-based resins to enhance specificity. Metal (IV) phosphates chemistry has revealed stronger coordination of metals like Zr4+ and Ti4+ with phosphate and carboxyl groups, which has been applied in methods such as Ti4+-IMAC. The preferential affinity of different metals for specific phosphopeptides has led to sequential enrichment strategies, such as Ti4+-IMAC which can enrich not only acidic residues but also multiple basic amino acid residues. Another example is the use of Ga3+-Fe3+-IMAC sequentially, where Ga3+ first enriches multiply phosphorylated peptides, then Fe3+ captures singly phosphorylated peptides. Extensive optimization of IMAC-based enrichment strategies has greatly improved their usability in targeted quantification.
(2) Metal Oxide Affinity Chromatography (MOAC)
MOAC enrichment is based on the principle of Lewis acid-base interactions, where metal oxides and hydroxides interact with the oxygen atoms of phosphate groups to form complexes. Metal oxides including titanium dioxide (TiO2) and zirconium dioxide (ZrO2) replace IMAC for selective enrichment of phosphopeptides. The most commonly used resin material currently is TiO2. The "offline method" using homemade/commercial titanium dioxide tips and the "online method" integrating titanium dioxide columns into multidimensional liquid chromatography setups have both been successfully applied. Buffer conditions used in MOAC significantly affect its selectivity for phosphopeptides. For example, adding 2.5-dihydroxybenzoic acid (DHB) to the loading and washing buffers can reduce non-specific binding of non-phosphorylated peptides to TiO2. Replacing DHB with lactic and β-hydroxypropionic acids, aliphatic hydroxy acids in the metal oxide chromatography (HAMMOC) modification, enhances the selectivity for phosphopeptides. Additionally, sequential elution on a pH gradient has been used to enhance enrichment efficiency. Because IMAC and MOAC have different affinities for phosphopeptides, combining the two enrichment strategies may be beneficial. IMAC and MOAC typically favor multiply phosphorylated and singly phosphorylated peptides, respectively, so both methods should be used simultaneously when mapping a comprehensive phosphorylation profile.
(3) Polymer-Based Metal Ion Affinity Capture (PolyMAC)
PolyMAC is another modification of IMAC, but it uses soluble nanopolymers instead of simple metal ions. Nanopolymers are dimers with multi-functionality of metal ions, capable of binding with phosphopeptides in solution. Subsequently, the PolyMAC-phosphopeptide complexes are recovered on solid phase by coupling with hydrazide agarose gel, followed by washing and elution of phosphopeptides. Compared to IMAC and TiO2 methods, the PolyMAC-Ti formulation has high recovery rates, reproducibility, and enrichment.
(4) Antibody-Based Phosphopeptide Enrichment
Antibodies against phosphoserine or phosphothreonine usually do not enrich phosphoproteins/peptides well. In contrast, anti-tyrosine phosphorylation antibodies, such as pY100, 4G10, and RC20, perform well in immunoprecipitation (IP) of tyrosine phosphorylated proteins and peptides for studying tyrosine signaling pathways. Due to the need for site-specific antibodies, immunoprecipitation is not suitable for broad-range studies of the phosphoproteome. However, the specificity and selectivity of antibodies provide a useful strategy for targeting specific low-abundance proteins, such as tyrosine phosphorylated proteins. Additionally, antibody-based and IMAC strategies can be combined in one workflow to cover the phosphoproteome more thoroughly.
(5) Strong Cation Exchange Chromatography (SCXC)
SCXC is the most commonly used LC for separating phosphopeptides. SCXC separates peptides based on their net charge state at low pH (pH 2-2.5). Because the C-terminus has Arg/Lys side chains and the N-terminus has amino groups, trypsin peptides typically have a net charge of 2+. Therefore, phosphopeptides elute from the SCXC column as part of the 1+ fraction before most unmodified trypsin peptides. However, the main limitation of SCXC fractionation is that multiply phosphorylated peptides and phosphopeptides with acidic amino acid residues often have a net negative charge state, and thus are not retained by the SCXC column. To avoid losing this large class of phosphopeptides, phosphopeptides that flow through must be collected and analyzed simultaneously. Because SCXC does not enrich to a high degree, this method is usually combined with affinity chromatography-based enrichment methods.
(6) Strong Anion Exchange Chromatography (SAXC)
SAXC is used for fractionating peptides based on negative charge. Under neutral pH conditions, phosphorylated and non-phosphorylated peptides are eluted at different salt concentrations. Additionally, singly and multiply phosphorylated peptides bind differently to SAXC materials. However, because net negative charge primarily depends on the number of acidic residues in the protein hydrolysates, the efficiency of separating non-phosphorylated and phosphorylated peptides is not high. Therefore, SAXC is less popular than SCXC.
(7) Anion and Cation Exchange Chromatography (ACEC)
To supplement the deficiencies of SCXC and SAXC, some studies use anion and cation exchange resins as another enrichment method before LC/MS analysis. The opposing charges in the mixed resin increase the retention time of acidic peptides, while shortening the retention time of basic and neutral peptides. Therefore, ACEC is the preferred method for enriching phage peptides, as it improves separation rates and recovery rates for phosphorylated and non-phosphorylated peptides.
(8) High pH Reverse Phase Liquid Chromatography (HpH RPLC)
HpH RPLC is another fractionation strategy based on hydrophobic interactions. It is conducted in a high pH environment, achieving orthogonal separation through a gradient of low to high pH in the mobile phase. Compared to SCXC and SAXC, HpH RPLC has higher resolution and can be coupled with low pH LC-MS/MS. In offline mode, RPLC can also select pooling of different gradient fractions, thus reducing the number of runs for instrument analysis. Pre-separation by RPLC before enriching phosphopeptides through IMAC or MOAC can broadly cover the phosphoproteome.
(9) Hydrophilic Interaction Liquid Chromatography (HILIC)
HILIC is another LC method for fractionating phosphopeptides. As its name suggests, HILIC has true orthogonal separation characteristics to reverse phase chromatography: it is based on a hydrophilic stationary phase and a hydrophobic organic mobile phase, separating peptides based on their overall hydrophilicity. Since phosphorylation alters the hydrophilicity of peptides, phosphopeptides bind strongly under HILIC conditions. Therefore, HILIC can more effectively fractionate phosphopeptides and enrich phosphopeptides based on the increased retention time of phosphopeptides in HILIC. In the near future, the application of HILIC in phosphoproteomics is likely to expand.
(10) Electrostatic Repulsion-Hydrophilic Interaction Chromatography (ERLIC)
ERLIC is a subset of HILIC that utilizes hydrophilic interactions and electrostatic forces. When there is a high percentage of organic mobile phase at low pH, hydrophilic interactions dominate over electrostatic forces, resulting in less chromatographic column repulsion (or retention) of phosphopeptides compared to non-phosphorylated peptides. However, lack of hydrophilic interactions causes electrostatic repulsion of phosphopeptides at low pH.
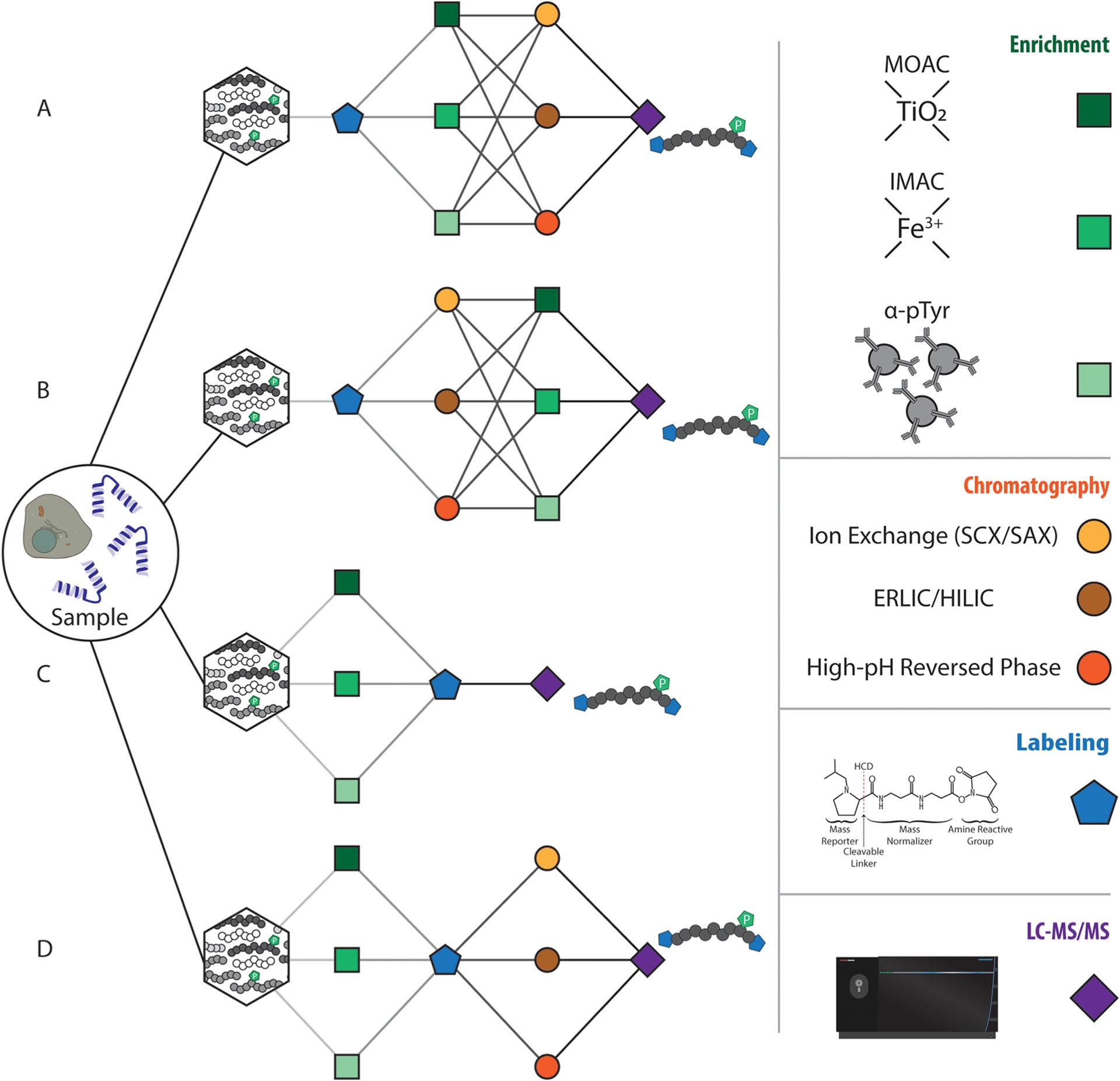
Figure 3. Analytical Scheme for Sample Handling in Phosphoproteomics [5]
2. Data Collection
Sample multiplexing typically focuses on using high-resolution instruments to differentiate nitrogen and carbon isotope defects. Due to the strong resolution capability and ease of use of the Orbitrap mass spectrometer, a substantial amount of work has been carried out. Generally, to resolve the nitrogen/carbon labels of TMT, a resolution of about 45,000 within the reported m/z range is required to accurately quantify all 16 channels in a TMTpro experiment. To achieve this resolution, each high-resolution scan takes about 100 milliseconds, equivalent to the 10 Hz scanning speed of the Orbitrap instrument. In contrast, ion traps and time-of-flight traps can reach scanning speeds of 50-100 Hz, but they cannot achieve the resolution required to distinguish all 16 reporting ions. The methods of sample multiplexing primarily use time or scan-based DDA methods and high-resolution MS2 (HRMS2) scans for identification and quantification. Although these methods are relatively fast, the ratio compression caused by TMT reduces the dynamic range of quantification. Therefore, a triplex scanning sequence (SPS-MS3) was developed, which uses synchronous precursor selection (SPS) in the MS2 stage to select TMT-containing peptide fragments, and a second round of fragmentation in the MS3 stage to release reporting ions for peptide quantification. This method significantly improves the quantitative accuracy of multiplex workflows but requires longer working cycles to achieve high-resolution MS3 scans. With the development of instruments, faster scanning speeds and stronger resolution capabilities will greatly enhance the depth and quantitative accuracy of phosphoproteomics.
Compared to unmodified peptides, the addition of phosphate active groups affects fragmentation behavior. To improve the analysis of phosphopeptides, researchers have employed various fragmentation techniques. Some fragmentation techniques evaluated for phosphopeptide analysis include: electron transfer dissociation (ETD), high-energy collision dissociation (ion beam CID, HCD), and ultraviolet photodissociation (UVPD), as well as their combinations (such as electron transfer/high-energy collision dissociation, EThcD), among which the HCD type fragmentation technique performs best in typical large-scale phosphoproteomics experiments. Although many of these methods are conducted in an unlabeled environment, they are readily applicable to isotope-labeled experiments.
The most common fragmentation strategy, especially in the case of isotope-labeled phosphopeptides, uses CID. CID includes two subclasses: ion trap CID and beam-type CID (i.e., high-energy CID or HCD). In the ion trap, peptides collide with inert gas (usually helium), transferring internal energy to the peptides. The transferred energy causes specific peptide ions to break at the most susceptible bonds, with phosphopeptide breakage occurring at the phosphate ester bonds. This type of breakage results in a neutral loss of precursor phosphate (98 Da). For many phosphopeptide ions, this fragmentation scheme is preferred, as only low-intensity b and y ions are observed. This neutral loss peak is usually the main peak in phosphopeptide CID tandem mass spectrometry, with only minor skeletal fragments observed. Methods combining MS3 fragmentation and pseudo-MS3 (multi-stage activation, MSA) are often used to increase the number of peptides available for sequence matching, thereby improving database searches. Additionally, in HCD, the collision is assisted by an electric potential, which accelerates peptide ions toward inert gas (usually nitrogen), causing higher energy collisions and the possibility of multiple fragment events per precursor. Thus, the intensity of the neutral loss precursor is greatly reduced, resulting in a richer and more complex fragmentation pattern.
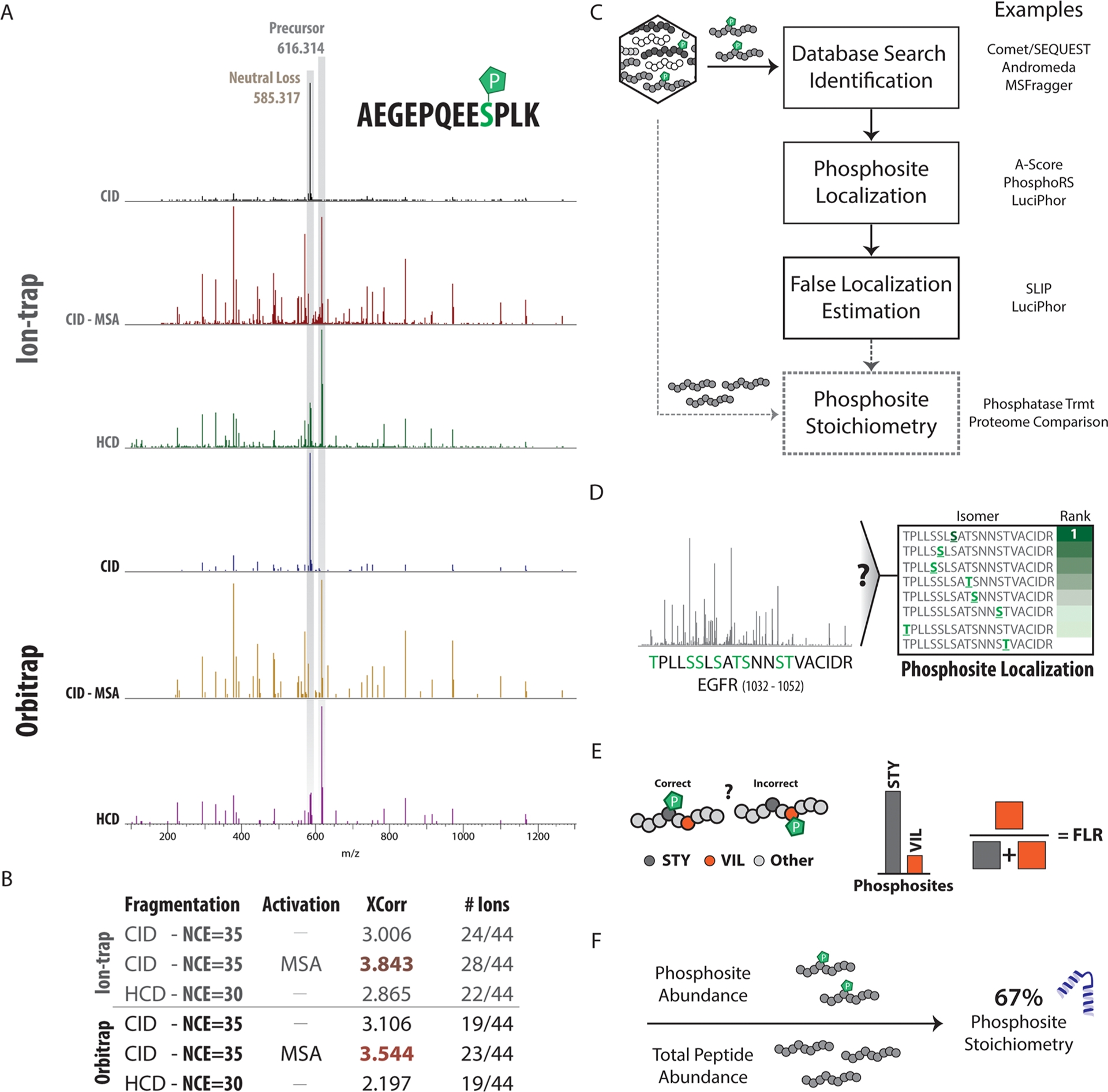
Figure 4. Collection and Data Analysis Strategies for Phosphoproteomics [5]
In contrast, electron capture dissociation (ECD) and ETD can preserve the labelable phosphate side chains, facilitating the identification of phosphopeptides. In ECD analysis, a low-energy electron is captured by a multiply-charged precursor ion. In ETD analysis, an electron is transferred from a free radical anion with low electron affinity to a peptide precursor cation. The recently developed EThcD technique combines ETD and HCD, producing rich MS/MS spectra with b/y and c/z ion pairs. Such spectra can achieve higher peptide sequence coverage and more confident localization of phosphorylation sites. However, these fragmentation schemes have slower scanning speeds, lower sensitivity, and are only effective for peptide segments with a charge state greater than 2+.
3. Data Analysis
The data analysis strategy for proteomics research is easily applicable to similar phosphoproteomic analyses. However, several additional phosphospecific steps are needed for accurate determination of phosphorylation sites, etc.
Open database retrieval techniques, used for identifying different post-translational modifications, involve searching spectra with very large parent ion tolerances. The quality number error of the scored peptides can then be evaluated to determine if the mass deviation is consistent with a specific post-translational modification. Software such as MSFragger, pFind, TagGraph, and MetaMorpheous have expanded the accessibility and efficiency of open database searches. These workflows can identify different modifications in the same analysis, including the identification of phosphorylation.
Site localization is a key computational challenge related to phosphoproteomic analysis. Since each serine, threonine, and tyrosine residue can potentially be phosphorylated, dynamic modifications lead to an increase in database size, thus making computational demands for phosphopeptide searches higher compared to unmodified peptides. Another complex issue is site localization, which usually requires measuring sequence-specific fragment ions, including complete phosphorylated molecules or fragment ions resulting from phosphate ester bond cleavage. Unlike general peptide identification, precursor mass and fragment alone are not the only factors needed for accurate identification. Instead, specific fragments are needed to determine the exact location of phosphorylation, and whether any potential phosphorylatable residues have any modifications. Most database search algorithms do not evaluate different peptide isomers, so regardless of the score or mass accuracy, they cannot distinguish between positional isomers of phosphorylated residues. For this purpose, several algorithms can help with site localization, including Mascot delta score, AScore, PTM score, PhosphoScore, Phosphinator, PhosphoRS, and LuciPHOr, etc.
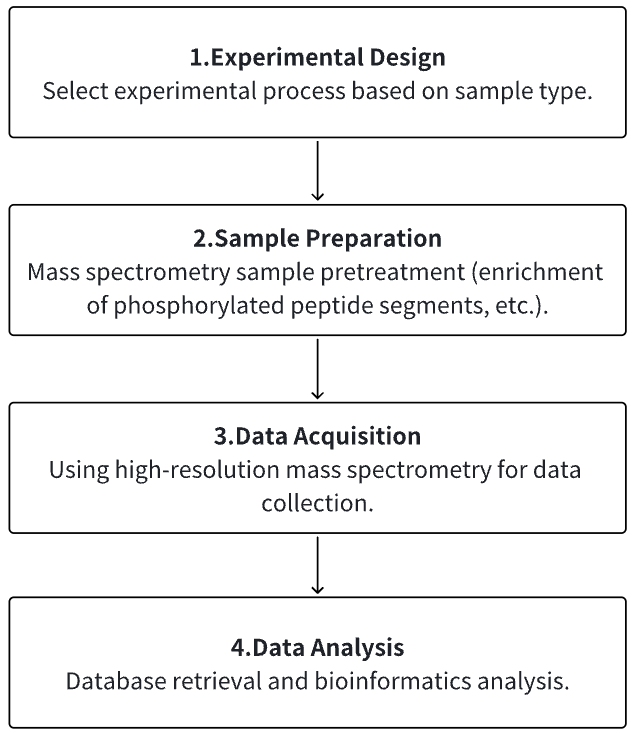
Analysis Workflow
1. According to the Experimental Requirements to Determine the Experimental Process
2. Mass Spectrometry Sample Preparation
3. High-resolution Mass Spectrometry Acquisition Data
4. Data Retrieval and Analysis
Service Advantages
1. Identification/Quantification/Modification Identification of Multi-Type Sample Source Proteins
2. High-confidence, High-precision Mass Spectrometry Detection
3. Comprehensive Bioinformatics Analysis
Sample Results
1. Quantitative Proteomics and Phosphoproteomics Analysis Reveal the Role of Death-Associated Protein Kinase 1 in Regulating Hippocampal Synapses
Death-associated protein kinase 1 (DAPK1) is a stress-responsive calcium/calmodulin (CaM)-regulated serine/threonine kinase actively involved in stress-induced cell death. Dysregulation of DAPK1 has been established in various neurological diseases such as epilepsy, Alzheimer's disease (AD), and Parkinson's disease (PD). Recent studies indicat the synaptic localization of DAPK1 in neurons, suggesting a potential role of DAPK1 in regulating synaptic structure and function. However, the key molecules and pathways through which DAPK1 influences synapses remain elusive. Studies using quantitative proteomics and phosphoproteomics analysis to compare protein expression and phosphorylation differences between wild-type (WT) and DAPK1 knockout (KO) mice hippocampal tissues have been conducted. Bioinformatics analysis of differentially expressed proteins and phosphoproteins showed enrichment of proteins involved in regulating synaptic function, cytoskeletal structure, and neurotransmission. Gene set enrichment analysis (GESA) highlighted altered presynaptic function in KO mice, including synaptic vesicle initiation and glutamate secretion. Furthermore, proteins with potential phosphorylation motifs of ERK and DAPK1 showed a high distribution in differentially phosphorylated proteins and were highly enriched in neuronal function-related pathways. Moreover, Western blot analysis confirmed the differences in expression of several proteins closely related to presynaptic tissue, dendrites, and calcium transmembrane transport between KO and WT mice, further confirming the potential involvement of DAPK1 in synaptic function regulation. Overall, the study provided molecular evidence for elucidating the physiological links between DAPK1 and neuronal function, aiding in clarifying the role of DAPK1 in the pathogenesis of neurodevelopmental and neurodegenerative diseases.
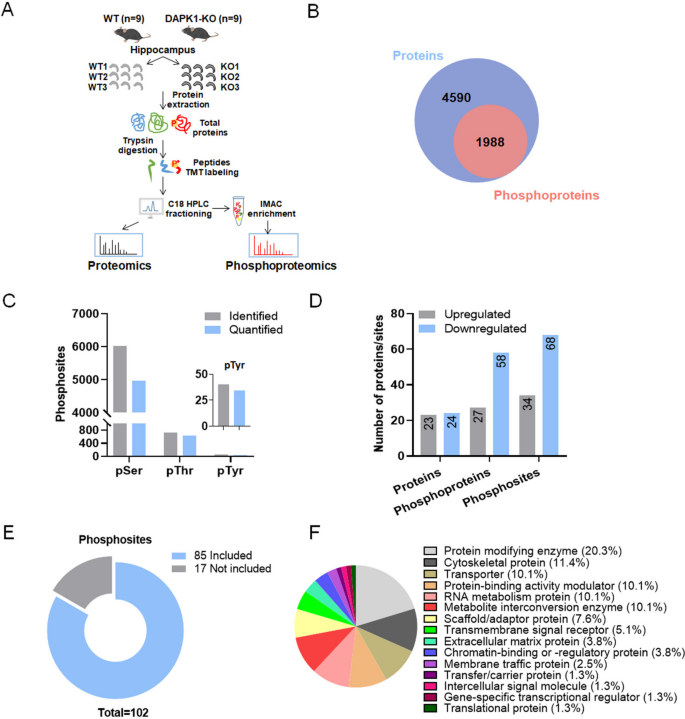
Figure 5. Quantitative/Phosphoproteomic Analysis of Hippocampal Tissues of WT and DAPK1-KO Mice [6]
2. Phosphoproteomics Reveals the Role of Serine/Threonine-Specific Protein Kinases in Cyanobacteria
Protein phosphorylation by serine/threonine protein kinases (Spk) is a widespread mechanism allowing cellular processes to adapt to changing environmental conditions. Studies have compared 11 of the 12 annotated Spks in the model cyanobacterium Synechocystis sp. PCC 6803, where mutants for each fully separated spk gene have been identified. Screening of the mutant collection revealed significant deviations in carbon metabolism, particularly in mutants of SpkB encoded by slr1697, which showed reduced growth rates under low CO2 or glucose presence, and a different glycogen accumulation pattern compared to WT. Changes in the ΔspkB proteome suggested alterations in cell surface properties, yet metabolic functions also changed. Phosphoproteomics analysis shows that two proteins in ΔspkB lack any phosphorylation, while phosphorylation of carboxysome-related protein CcmM decreased, and phosphorylation of allophycocyanin alpha subunit ApcA increased. Moreover, compared to WT, mutants showed lower phosphorylation levels of PII protein, confirmed by Western blotting, indicating a significant delay in PII phosphorylation when cells were transferred from nitrate-containing to nitrate-free media. Findings suggested that SpkB was an important regulator in Synechocystis, involved in the phosphorylation of PII protein and other proteins.
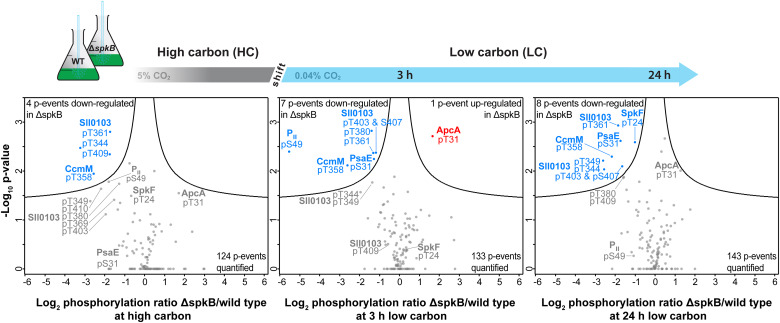
Figure 6. Quantitative Comparison of Phosphoproteomic Changes in ΔspkB Mutants and WT under Different Conditions [7]
3. Phosphorylation of MAP 1A Regulates Excessive Phosphorylation of Tau in Alzheimer's Disease Models
Excessive phosphorylation of Tau protein is one of the key pathological features of Alzheimer's disease (AD). Therefore, studying the mechanisms behind Tau hyperphosphorylation is crucial for exploring the pathogenesis of AD-related neuronal damage. Studies have established an AD rat model and conducted quantitative phosphoproteomics and proteomics to identify proteins. It showed that a reduction phosphorylation levels of microtubule-associated protein 1A (MAP 1A) in the model group has been observed. Western blot confirmed changes in MAP 1A in SD rats, APP/PS1 transgenic mice, and cellular AD models. To further study the molecular mechanisms by which recombinant MAP 1A phosphorylation affected Tau phosphorylation, siRNA-MAP 1A interference and protein immunoprecipitation assays were conducted in AD cell models. The research found that the downregulation of p-MAP 1A associated with upregulation of p-Tau was due to their changed binding affinity with CDK5, and MAP 1A can competitively bind with CDK5 to antagonize Tau phosphorylation, enhancing self-phosphorylation. This results in neuronal protection and reduced AD tissue damage aided in a better understanding of the pathogenesis of AD.
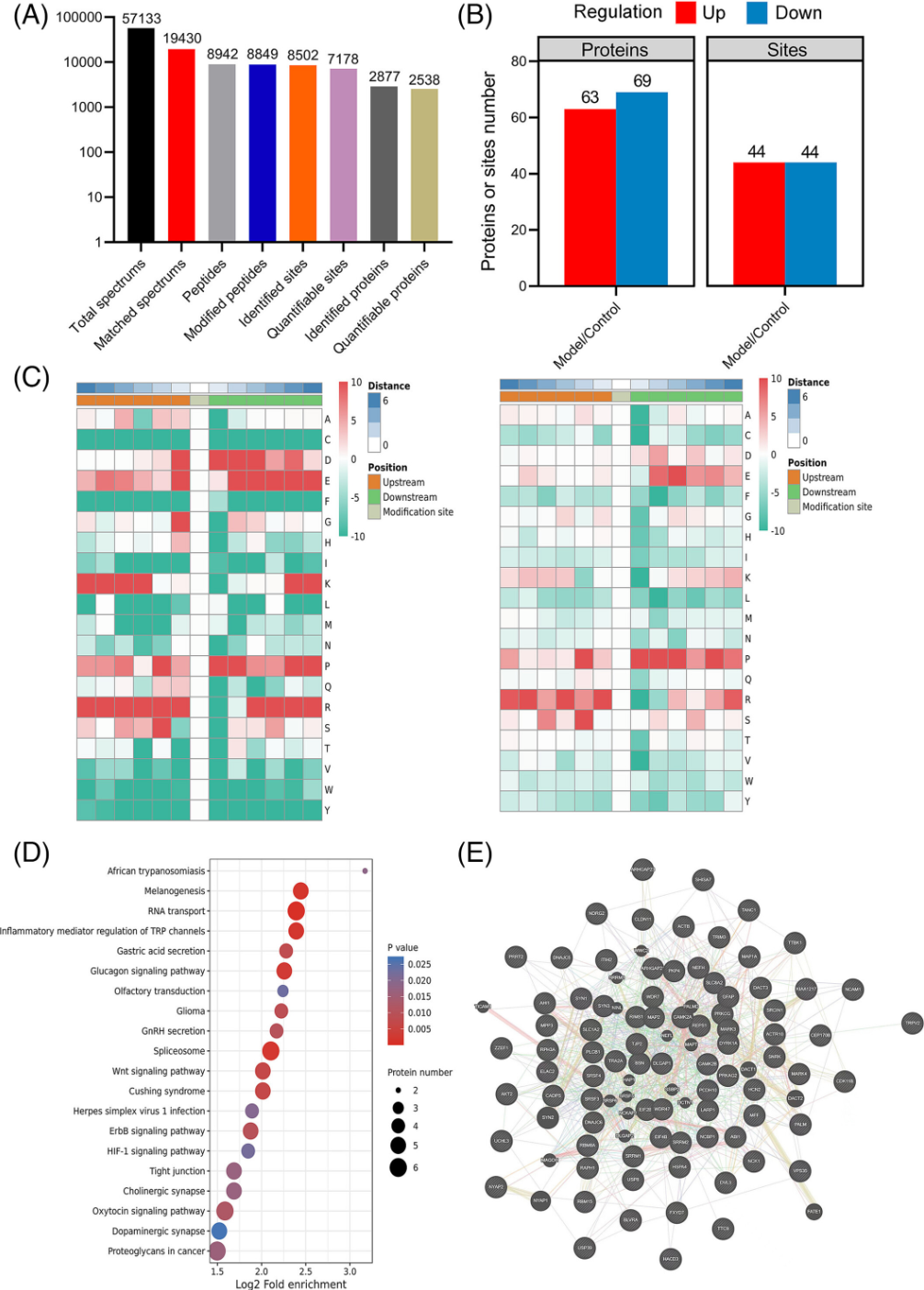
Figure 7. Quantitative Phosphoproteomics and Proteomics Identification of AD Rat Proteins [8]
Sample Submission Requirements
1. Protein Purity >90%
2. Try to Avoid Impurity Contamination
Services at MtoZ Biolabs
1. Experimental Procedure
2. Relevant Mass Spectrometry Parameters
3. Raw Data
4. Data Analysis Reports
Applications
1. Application of Quantitative Phosphoproteomics in Cancer
Quantitative phosphoproteomics can identify aberrantly activated signaling pathways and therapeutic targets in cancer. It also reveals downstream effectors of mutated kinases, providing opportunities to understand the molecular mechanisms leading to tumor development. Identifying cellular targets of kinase inhibitors in cancer and the off-target effects of these inhibitors are some other interesting applications of this strategy.
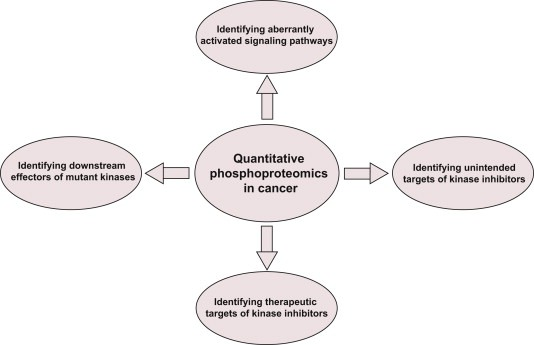
Figure 8. Application of Quantitative Phosphoproteomics in Cancer [4]
2. Phosphoproteomics Methods for Identifying Phosphatase and Kinase Substrates
Protein phosphorylation is a ubiquitous post-translational modification, controlled by the opposing activities of protein kinases and phosphatases, which regulate various biological processes across all kingdoms of life. A major challenge in understanding the phosphoregulatory network is the clear identification of kinase and phosphatase substrates. Liquid chromatography coupled with mass spectrometry (LC-MS/MS) and related phosphoproteomics tools can globally investigate phosphoproteome changes in response to signaling events or perturbantion in phosphoregulatory network components. LC-MS/MS can be applied in various types of phosphoproteomics experiments to study kinase/phosphatase functions and identify candidate substrates. Research methods can be divided into three categories: the first involves disrupting in vivo target kinase/phosphatase activity and measuring subsequent changes in the phosphoproteome, thereby revealing target-dependent specific physiological phosphorylation sites. The second includes methods dependent on affinity capturing enzyme-substrate complexes or in vivo proximity-labeled substrates from biological systems. The third covers in vitro assays using substrate pools derived from cell extracts or synthetic peptide libraries.
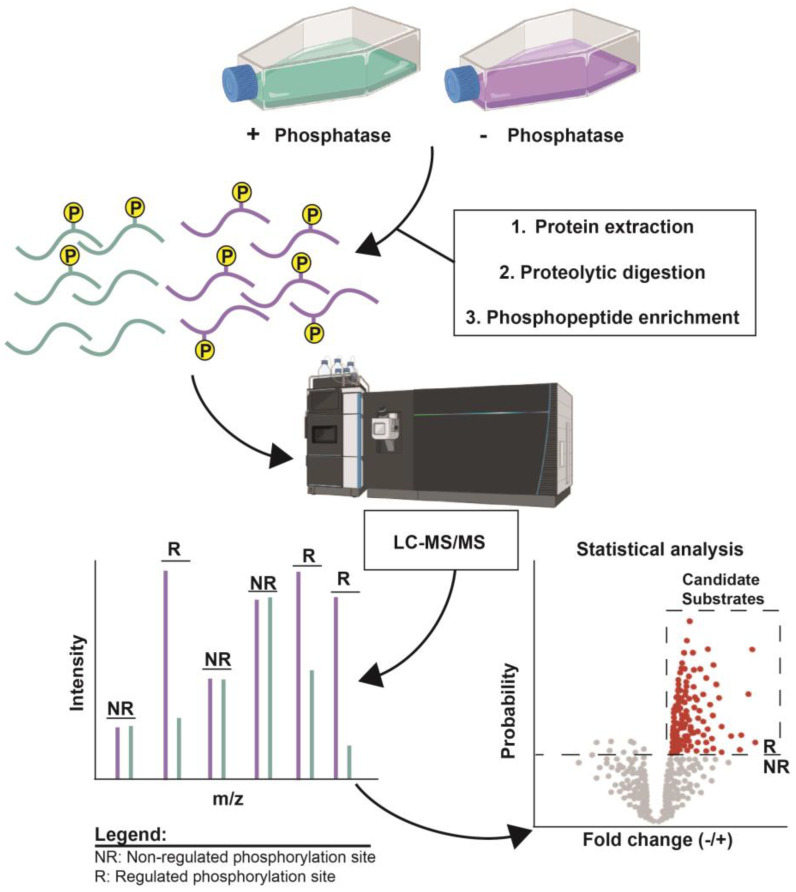
Figure 9. Examples of Identifying Candidate Phosphatase Substrates by Altering Target Enzyme Activity [9]
FAQ
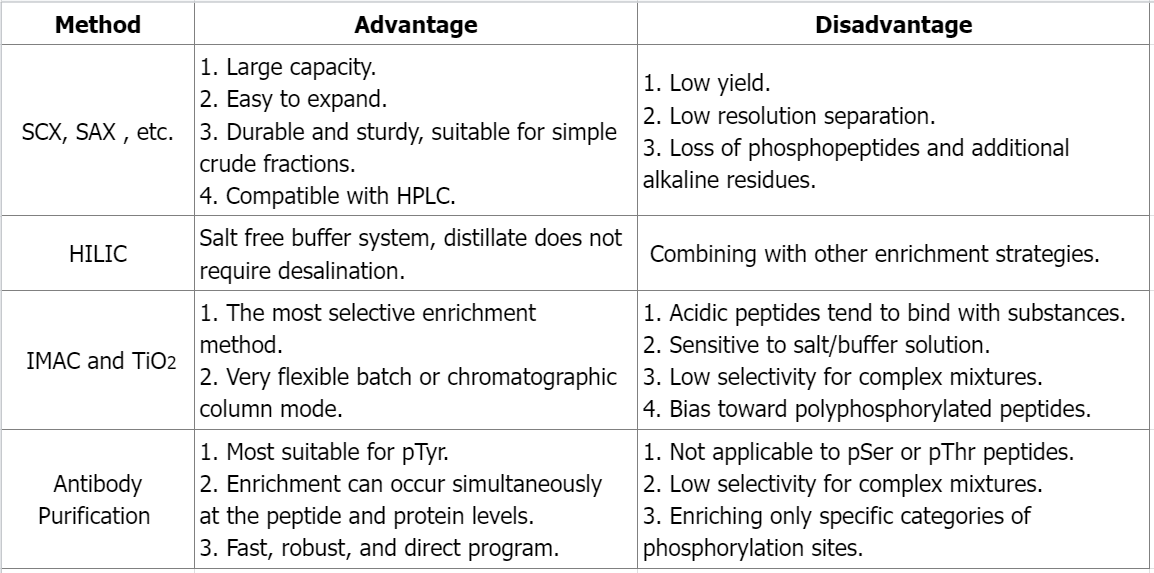
Q1: What are the advantages and disadvantages of different phosphate peptide enrichment methods?
References
[1] Cox J, Mann M. Quantitative, high-resolution proteomics for data-driven systems biology. Annu Rev Biochem. 2011;80:273-99. doi: 10.1146/annurev-biochem-061308-093216. PMID: 21548781.
[2] Ahire D, Kruger L, Sharma S, Mettu VS, Basit A, Prasad B. Quantitative Proteomics in Translational Absorption, Distribution, Metabolism, and Excretion and Precision Medicine. Pharmacol Rev. 2022 Jul;74(3):769-796. doi: 10.1124/pharmrev.121.000449. PMID: 35738681; PMCID: PMC9553121.
[3] Derouiche A, Cousin C, Mijakovic I. Protein phosphorylation from the perspective of systems biology. Curr Opin Biotechnol. 2012 Aug;23(4):585-90. doi: 10.1016/j.copbio.2011.11.008. Epub 2011 Nov 24. PMID: 22119098.
[4] Harsha HC, Pandey A. Phosphoproteomics in cancer. Mol Oncol. 2010 Dec;4(6):482-95. doi: 10.1016/j.molonc.2010.09.004. Epub 2010 Sep 26. PMID: 20937571; PMCID: PMC3030978.
[5] Paulo JA, Schweppe DK. Advances in quantitative high-throughput phosphoproteomics with sample multiplexing. Proteomics. 2021 May;21(9):e2000140. doi: 10.1002/pmic.202000140. Epub 2021 Mar 30. PMID: 33455035; PMCID: PMC8209658.
[6] Tian Y, Zheng X, Li R, Hu L, Shui X, Wang L, Chen D, Lee TH, Zhang T. Quantitative Proteomic and Phosphoproteomic Analyses Reveal a Role of Death-Associated Protein Kinase 1 in Regulating Hippocampal Synapse. Mol Neurobiol. 2023 Sep 30. doi: 10.1007/s12035-023-03674-4. Epub ahead of print. PMID: 37775722.
[7] Barske T, Spät P, Schubert H, Walke P, Maček B, Hagemann M. The Role of Serine/Threonine-Specific Protein Kinases in Cyanobacteria - SpkB Is Involved in Acclimation to Fluctuating Conditions in Synechocystis sp. PCC 6803. Mol Cell Proteomics. 2023 Oct 4;22(11):100656. doi: 10.1016/j.mcpro.2023.100656. Epub ahead of print. PMID: 37797745.
[8] Cai B, Shao N, Ye T, Zhou P, Si W, Song H, Wang G, Kou J. Phosphorylation of MAP 1A regulates hyperphosphorylation of Tau in Alzheimer's disease model. Neuropathol Appl Neurobiol. 2023 Oct;49(5):e12934. doi: 10.1111/nan.12934. PMID: 37705167.
[9] DeMarco AG, Hall MC. Phosphoproteomic Approaches for Identifying Phosphatase and Kinase Substrates. Molecules. 2023 Apr 24;28(9):3675. doi: 10.3390/molecules28093675. PMID: 37175085; PMCID: PMC10180314.
MtoZ Biolabs, an integrated chromatography and mass spectrometry (MS) services provider.
Related Services
Quantitative Tyrosine Phosphoproteomics Analysis Service
How to order?

