





De Novo Sequencing: Principles, Advantages, and Applications in Proteomics Research
-
target protein identity or expected size, if known
-
sample purity estimate or gel image
-
organism or expression system background
-
known modifications or expected sequence differences
-
whether full-length coverage or regional confirmation is required
Introduction
Many proteomics projects depend on matching MS/MS spectra to entries in a protein sequence database. Database search works well when the sample comes from a well-annotated organism and the protein sequence is already known. However, proteomics increasingly involves proteins that cannot be explained by an existing reference. Novel proteins, unsequenced organisms, engineered antibodies, recombinant constructs, splice variants, and sequence mutations all create gaps that database search cannot fill on its own.
De novo sequencing addresses this gap by deriving peptide sequence directly from tandem mass spectra. Instead of asking which known protein best explains a spectrum, the method reconstructs the amino acid sequence from fragment ion patterns. This makes the approach valuable when the biological question depends on sequence information that is missing, incomplete, or intentionally modified. In practical proteomics workflows, the method is often combined with database search, Edman degradation, gene sequencing, or peptide mapping to build a complete sequence report.
Related Services
| Research Need | Recommended Service Direction |
| Sequence proteins without a reliable database match | De Novo Sequencing Service |
| Confirm full protein sequence by mass spectrometry | De Novo Protein Sequencing Service |
| Sequence unknown or novel antibodies | De Novo Antibody Sequencing Service |
| Analyze proteins with incomplete reference coverage | Unknown Proteins Sequencing Service |
| Support broader sequence and modification analysis | Protein Sequencing Service by Mass Spectrometry |
For projects where database search returns weak matches or no confident identification, MtoZ Biolabs can help evaluate whether de novo protein sequencing, antibody de novo sequencing, peptide mapping, or a combined MS-based sequence workflow fits the sample type and reporting goal.
What Is De Novo Sequencing?
De novo sequencing is a mass spectrometry-based approach that determines peptide sequence from MS/MS fragmentation data without relying on a prior protein database match. A peptide is ionized and fragmented in the mass spectrometer. The resulting b-ions, y-ions, and related fragment series reflect mass differences between adjacent residues. Software or expert interpretation uses these mass differences to infer the amino acid order.
At the protein level, de novo protein sequencing usually means assembling multiple de novo- derived peptides into a longer contiguous sequence. A single peptide may be enough for a short region confirmation. Full-length coverage typically requires complementary proteases, fractionation, higher MS depth, and manual review of ambiguous residues. The method is therefore both a peptide-level and protein-level strategy, depending on project depth and sample complexity.
The method is not the same as database identification. Database search asks whether a known sequence can explain the spectrum. Sequence-from-spectrum analysis asks what sequence the MS/MS data itself supports. That distinction matters whenever the true sequence may differ from public databases.
How the Method Works
A typical workflow starts with protein purification or enrichment, followed by enzymatic digestion. Trypsin is common, but alternative proteases such as chymotrypsin, Glu-C, or Lys-C can improve coverage when tryptic sites are sparse or blocked. Peptides are separated by liquid chromatography to reduce ion suppression and improve MS/MS quality.
During MS/MS acquisition, selected precursor ions are isolated and fragmented. The fragment pattern encodes residue-level information. Sequence assembly algorithms score possible amino acid strings against the observed ions. Higher mass accuracy improves confidence in distinguishing residues with similar mass, such as isoleucine and leucine.
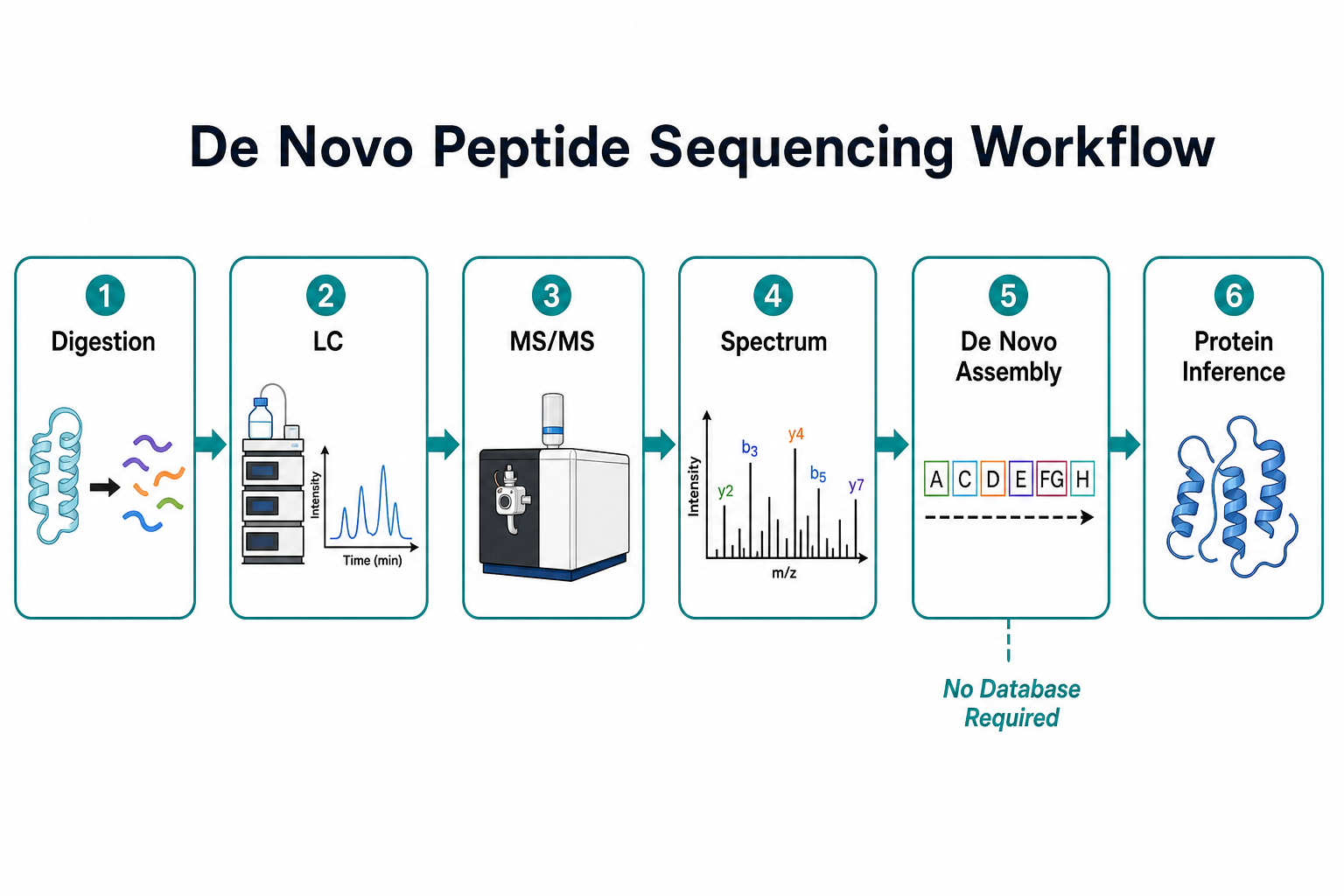
Figure 1. MS/MS workflow for sequence-from-spectrum analysis
Successful sequence assembly depends on spectrum quality. Strong precursor intensity, clean fragmentation, sufficient sequence coverage within the peptide, and low chemical noise all improve assembly accuracy. Post-translational modifications, missed cleavages, and in-source fragmentation can complicate interpretation. Raw spectrum review remains important even when automated tools generate a candidate sequence.
Overlapping peptides from multiple digests help resolve ambiguous residues and support longer sequence reconstruction. Peptide de novo sequencing becomes more informative when different enzymes generate redundant coverage across the same region.
Comparison with Database Search
Database search remains the default strategy for large-scale proteomics because it is efficient and scalable when reference proteomes are complete. Sequence-from-spectrum analysis becomes essential when the reference is incomplete, incorrect, or absent. Engineered proteins, hybrid sequences, venoms, microbiota from poorly annotated species, and proprietary biologics are common examples.
Database search can also miss sequence variants if the variant is not present in the database. A single amino acid substitution may prevent a confident peptide-spectrum match even when the MS/MS spectrum is high quality. MS/MS-derived sequencing can reveal the variant-containing peptide directly, making it useful for mutation analysis, biosimilar comparison, and quality assessment of recombinant proteins.
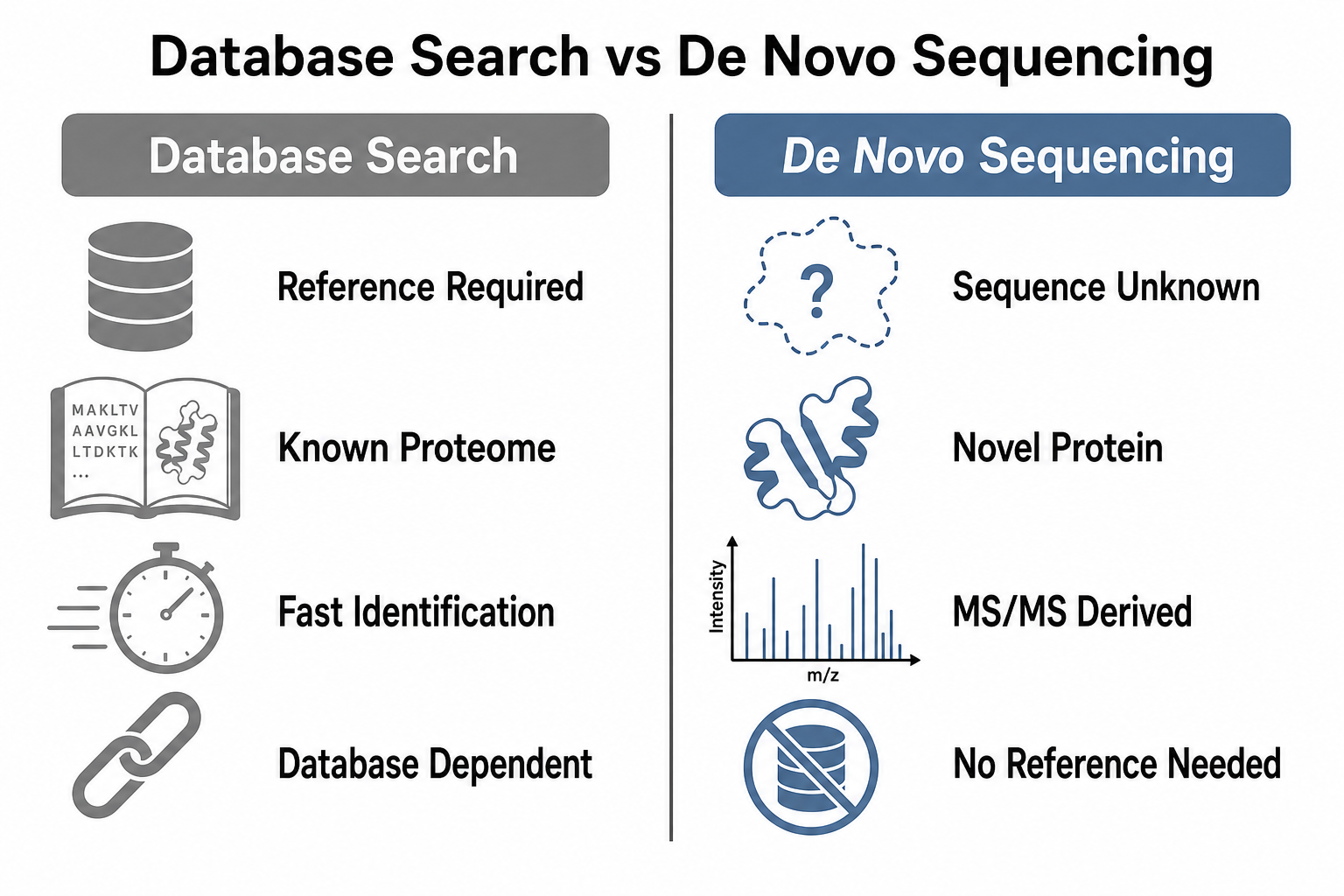
Figure 2. Database search and sequence-from-spectrum analysis answer different questions
In practice, many high-confidence projects use both approaches. Database search can map the majority of peptides in a well-characterized background proteome. De novo analysis can then focus on unmatched spectra, low-scoring peptides, or regions where biological evidence suggests a novel or modified sequence. This hybrid strategy is common in antibody sequencing, unknown protein identification, and biopharmaceutical characterization.
Core Advantages
The method offers several strengths that database-dependent workflows cannot fully replace.
1. Sequence Discovery Without a Reference
The method can support identification when no suitable database entry exists.
2. Variant and Mutation Detection
MS/MS-derived sequence can reveal substitutions, insertions, deletions, and unexpected modifications.
3. Support for Engineered Proteins
Recombinant constructs, fusion proteins, and therapeutic antibodies can be analyzed even when the exact construct is proprietary.
4. Complement to Edman and Gene Sequencing
MS-based sequence can validate N-terminal regions, internal peptides, and expressed protein products.
5. Useful for Low-database Organisms
Studies on non-model species benefit when genomic annotation lags behind MS capability.
These advantages make the approach especially relevant when sequence confirmation affects publication, patent support, clone selection, or regulatory documentation.
Current Limitations
The approach also has clear boundaries. Low-quality spectra, insufficient fragmentation, and highly homologous repeat regions can reduce confidence. Isoleucine and leucine are often indistinguishable by mass alone, so orthogonal evidence may be needed. Complex mixtures can produce chimeric spectra that are difficult to interpret at the peptide level.
Computational interpretation adds another layer of complexity. Different algorithms may propose slightly different sequences for the same spectrum. Manual curation is often required for high- stakes reporting. Compared with large-scale database search, de novo analysis is more labor- intensive and less suited to unattended high-throughput identification across entire proteomes.
Sample preparation limits apply as well. Heavy contamination, poor digestion, chemical modifications, and low protein amount can all reduce usable MS/MS data. The workflow is strongest when the target protein or peptide fraction is enriched and the MS method is optimized for sequence coverage rather than only protein identification count.
Typical Applications in Proteomics
The method is widely used when sequence information has direct scientific or commercial value.
1. Unknown Protein Identification
When a purified band or enriched protein cannot be matched confidently to a database, de novo protein sequencing can provide candidate sequence for cloning, functional study, or homology analysis.
2. Antibody Sequencing
Therapeutic and research antibodies may require full variable region sequence when hybridoma sequence data are incomplete or unavailable. De novo antibody sequencing combines peptide-level MS interpretation with overlap assembly across light and heavy chains.
3. Protein Variant and Mutation Analysis
Substitutions introduced by expression systems, cell lines, or manufacturing changes may be detected as de novo-derived peptides that do not align with the expected reference.
4. Biopharmaceutical Characterization
Sequence confirmation supports comparability studies, biosimilar assessment, and primary structure documentation for recombinant proteins and peptides.
5. Non-model and Environmental Proteomics
Species with incomplete genomes or metaproteomics samples may yield high-quality spectra that database search cannot explain. MS/MS-derived sequencing helps recover biologically informative sequence from those datasets.
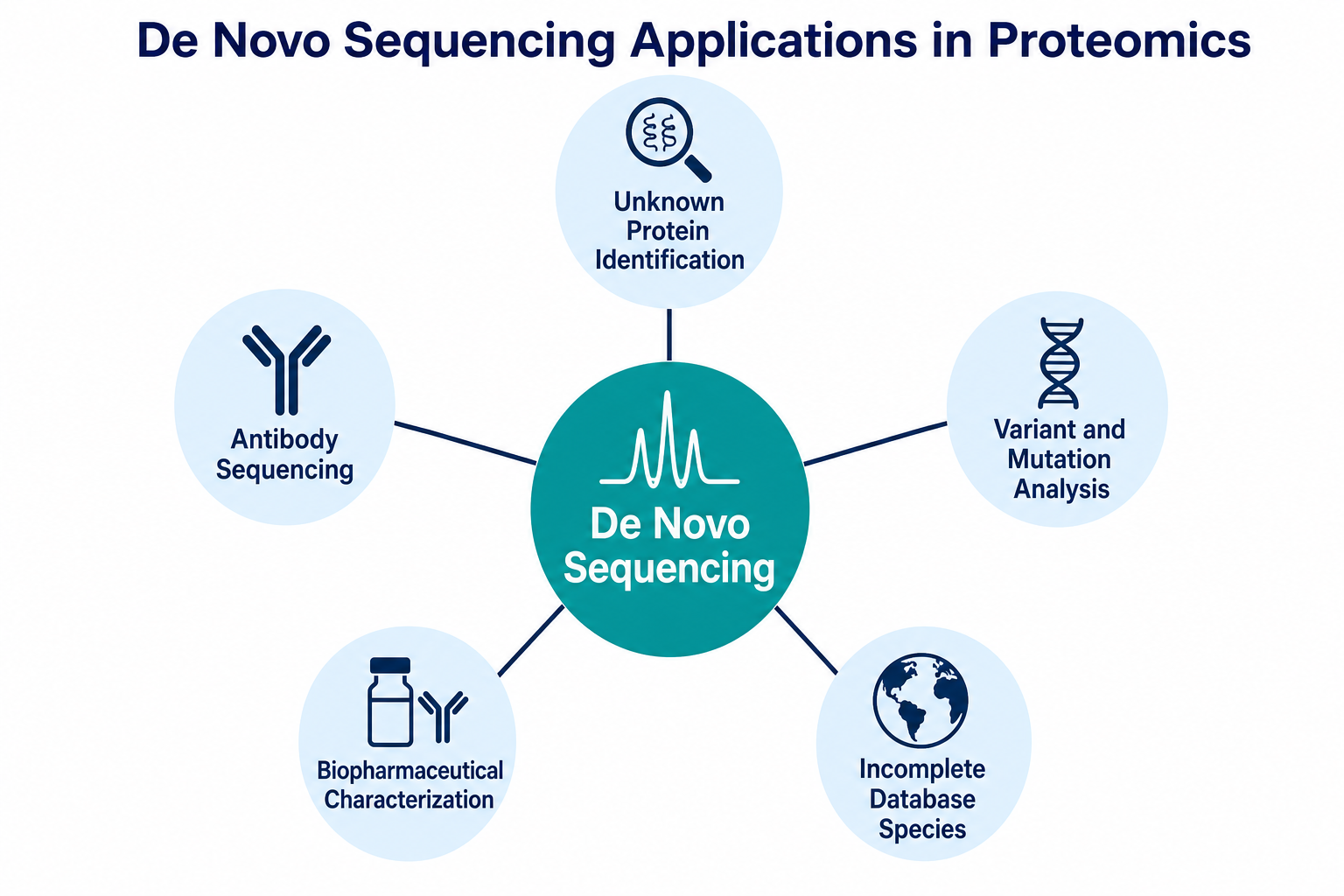
Figure 3. Common proteomics applications of the method
Across these applications, the reporting goal should be defined early. Some projects need a short confirmed peptide region. Others require maximal sequence coverage of a full protein or antibody chain. The required depth influences digestion strategy, instrument time, and validation design.
Sample and Data Requirements
Strong de novo results usually depend on sample quality more than software choice alone. Purified or highly enriched protein samples generally perform better than complex crude lysates. For gel-based samples, clean excision and minimal keratin contamination improve interpretability.
Useful project inputs include:
High-resolution MS/MS data with good fragment ion series are preferred. Multiple enzymes, replicate runs, and overlapping peptides increase the chance of unambiguous assembly. When the project allows, combining the method with Edman N-terminal analysis, PCR-based antibody sequencing, or peptide mapping can strengthen the final sequence report.
Future Outlook
The method is likely to remain a specialized but essential tool in proteomics rather than a wholesale replacement for database search. Instrument improvements, higher scan speed, better fragmentation methods, and more accurate mass analyzers continue to improve spectrum quality.
The most promising direction is integrated sequencing strategy. Database search, sequence-from- spectrum analysis, targeted MS, chemical derivatization for isoleucine and leucine distinction, and orthogonal methods can be combined into a single reporting workflow. This integrated model fits antibody discovery, biologics development, and any project where sequence certainty matters more than protein identification count alone.
Frequently Asked Questions
1. What is de novo sequencing in proteomics?
The process determines peptide or protein sequence directly from MS/MS spectra without requiring a prior database match. The sequence is inferred from fragment ion patterns produced during tandem mass spectrometry.
2. When should researchers use this approach instead of database search?
Researchers should consider sequence-from-spectrum analysis when the protein is unknown, the database is incomplete, the sequence is engineered or proprietary, or a variant is suspected but not represented in reference data.
3. Can the method determine full protein sequence?
Yes, in favorable cases. Full de novo protein sequencing usually requires multiple peptides, overlapping coverage, complementary digestion, and manual review. Low-abundance proteins or highly complex mixtures may support only partial sequence coverage.
4. Is this approach the same as Edman degradation?
No. Edman degradation determines N-terminal sequence chemistry step by step. MS/MS-based sequencing uses fragment patterns and can analyze internal peptides from digested proteins. The methods are often complementary.
5. What are the main limitations?
Main limitations include poor spectrum quality, leucine and isoleucine ambiguity, complex mixtures, chimeric spectra, and higher analysis effort compared with routine database search.
Conclusion
De novo sequencing enables proteomics to move beyond reference-dependent identification. By deriving sequence from MS/MS data, the method supports unknown protein identification, antibody sequencing, variant analysis, biopharmaceutical characterization, and studies in poorly annotated biological systems. Its value is strongest when sample preparation, instrument performance, and reporting goals are aligned from the start.
For projects that need reliable sequence evidence beyond database matching, contact MtoZ Biolabs to discuss de novo sequencing, antibody sequencing, or an integrated MS-based sequence analysis workflow.
How to order?

