





De Novo Protein Sequencing vs Database-Based Identification: How to Choose
-
the background proteome is complex but one protein is the real focus
-
an expected construct must be checked for undocumented differences
-
a variant peptide is suspected but absent from the reference database
-
only a small region requires sequence-level proof
-
Is a reliable reference sequence available for this exact protein?
-
Does the project require discovery of unknown sequence or confirmation of a known design?
-
Is the sample enriched enough for sequence assembly?
-
Is full-length coverage required, or is partial confirmation acceptable?
-
Will the report be used for internal QC, publication, or sequence-driven cloning?
Introduction
Protein sequencing projects often reach a decision point where two LC-MS/MS strategies appear equally plausible. Database-based identification matches experimental spectra to known protein entries and is efficient when the reference is reliable. De novo protein sequencing derives sequence information directly from peptide fragmentation data when the correct reference is missing, incomplete, or untrustworthy. The wrong choice can waste sample, delay cloning or QC decisions, and produce a report that looks complete but does not answer the real biological question.
The comparison is not about which method is more advanced. Database-based identification asks which known sequence best explains the data. De novo protein sequencing asks what sequence the data themselves support. A recombinant batch with an undocumented variant, a gel band from a poorly annotated source, or a proprietary construct may fail database matching even when the LC-MS/MS data are strong. Conversely, a well-characterized human cell-line sample may not need de novo analysis at all. The best workflow depends on reference quality, project goal, and the level of sequence proof required.
Related Services
| Service Area | Recommended Service |
|---|---|
| De novo protein sequencing | |
| Database-based identification | |
| Reference confirmation | |
| Unknown protein sequencing | |
| Full-length sequence recovery | |
| Broader MS sequencing support |
Researchers unsure which route fits their sample can consult MtoZ Biolabs to compare reference availability, coverage goals, and reporting needs before committing LC-MS/MS time.
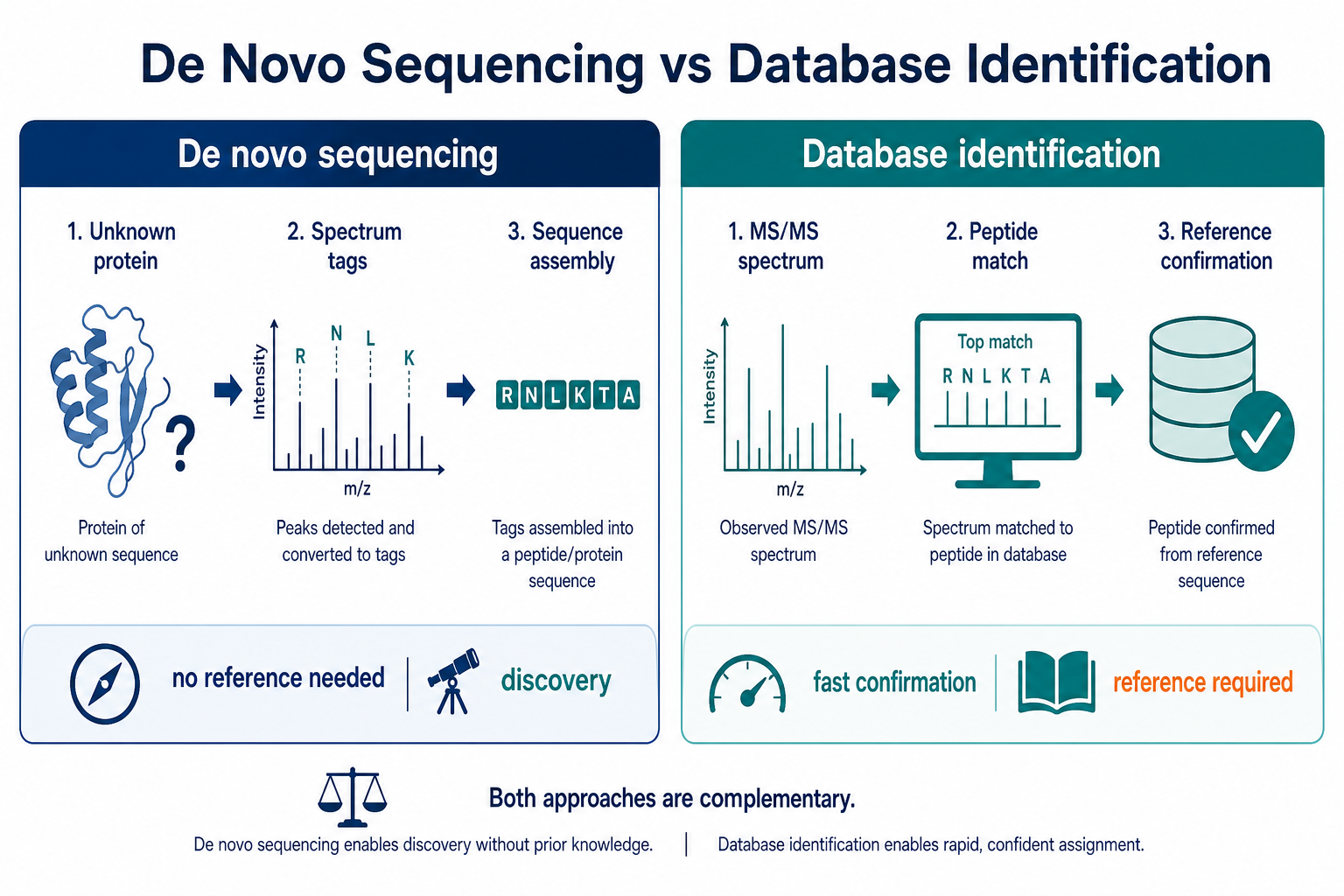
Figure 1. De novo protein sequencing supports unknown or divergent sequences, while database-based identification is optimized for reference-backed confirmation.
When Researchers Face This Decision
This comparison usually appears in a few recurring scenarios.
A purified protein band may produce peptides, but database search returns weak or ambiguous matches. A recombinant product may show partial agreement with the expected construct, suggesting a variant or processing event. An unknown protein from a non-model organism may lack a suitable reference proteome. A legacy sample may have no genetic record, making direct sequence recovery the only practical option. In each case, the key question is whether the study needs to confirm a known sequence or discover the sequence from the sample itself.
Choosing too early can create downstream rework. If database-based identification is used when the reference is wrong, the project may stop at a false match. If de novo protein sequencing is used when a strong reference already exists, the project may spend more time and sample than necessary. A short feasibility review before digestion and LC-MS/MS often prevents this mismatch.
Four Comparison Dimensions That Matter Most
A useful comparison should focus on decision variables rather than generic method descriptions.
Reference availability. Database-based identification depends on a complete and accurate sequence database. De novo protein sequencing is designed for cases where the correct sequence cannot be assumed in advance.
Study goal. Broad protein identification favors database search. Unknown protein characterization, variant detection, antibody sequencing, and sequence confirmation for proprietary proteins often require de novo analysis or a hybrid workflow.
Throughput and scale. Database-based identification scales well across complex samples and large peptide lists. De novo protein sequencing is more resource-intensive and is usually targeted to enriched proteins, unmatched spectra, or sequence-critical regions.
Required evidence level. Database matching can confirm identity when the reference is trustworthy. De novo assembly can provide primary structure evidence but often requires expert review, overlapping peptides, and orthogonal confirmation for high-confidence reporting.
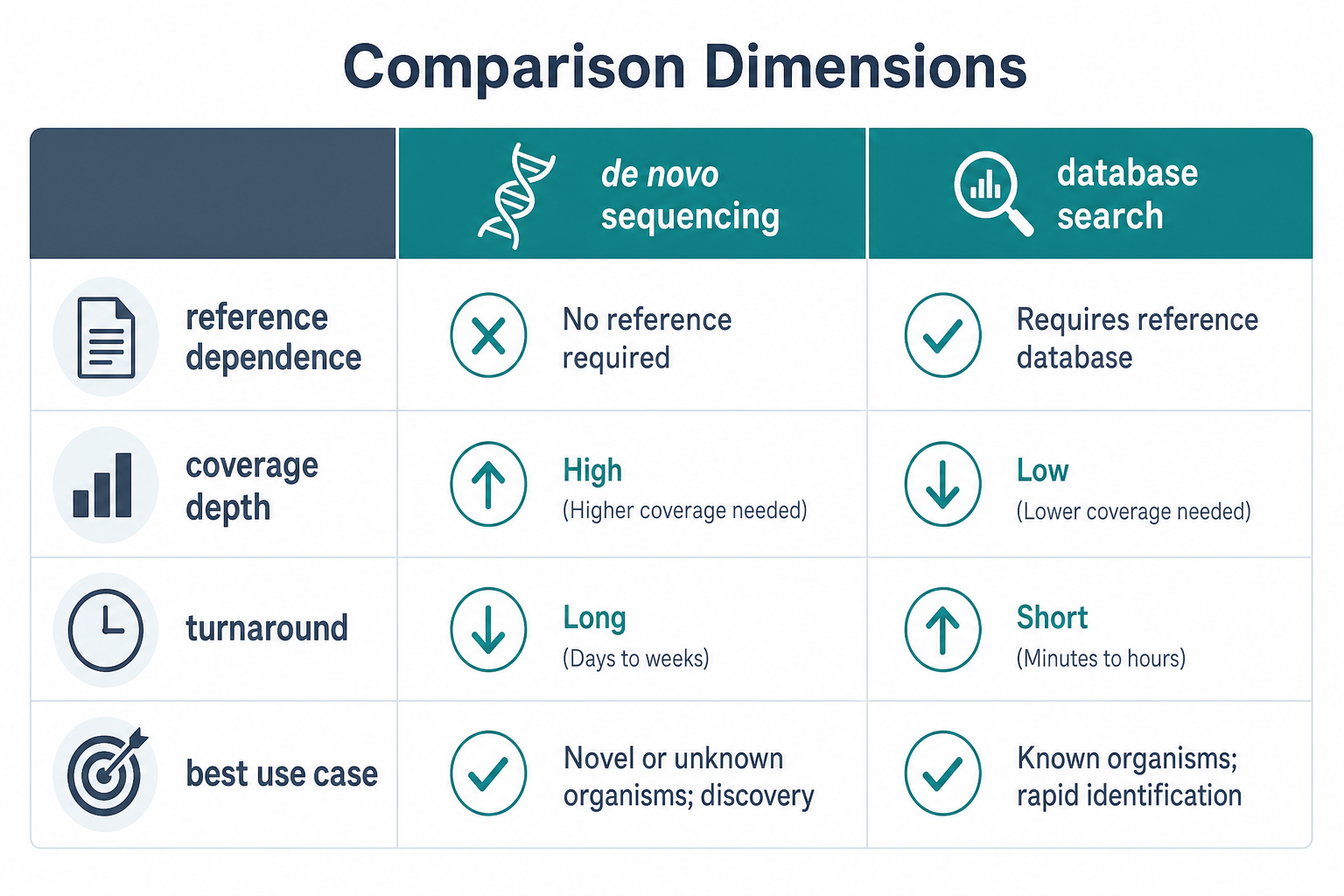
Figure 2. Reference dependence, coverage depth, turnaround, and use case fit are the main dimensions for method selection.
How Database-Based Identification Works
Database-based identification matches experimental MS/MS spectra to in silico fragment ions generated from a protein sequence database. Search engines score peptide-spectrum matches using precursor mass accuracy, fragment coverage, enzyme specificity, modification parameters, and false discovery rate control.
The method performs well when the database represents the sample accurately. It is the default choice for many proteomics projects involving model organisms, human samples, standard cell lines, and well-annotated expression systems. It supports protein identification, quantification, and modification mapping at scale.
The main weakness appears when the measured sequence is absent from the database. Missing isoforms, undocumented mutations, expression construct differences, fusion tags, signal peptide processing, and cross-species homology gaps can all reduce match quality. In those cases, poor results may reflect reference limitations rather than poor MS data.
How De Novo Protein Sequencing Works
De novo protein sequencing interprets peptide fragmentation patterns without requiring a prior database match. Analysts derive sequence tags from b-ions and y-ions in MS/MS spectra, then assemble overlapping peptides into longer regions and, when coverage allows, a protein-level sequence.
This approach is valuable for unknown proteins, proprietary constructs, antibody variable regions, venoms, environmental samples, and any project where the measured sequence may differ from public references. It is strongest when the target protein is enriched, spectra are high quality, and the project requires direct primary structure evidence.
The method is less efficient for whole-proteome screening of complex lysates without prioritization. It also depends on spectrum quality, manual review, and overlap validation. Ambiguous residues, homologous regions, and post-translational modifications can limit confidence if the workflow is not designed carefully.
Side-by-Side Comparison
| Dimension | De Novo Protein Sequencing | Database-Based Identification |
|---|---|---|
| Reference requirement | Low; works without a trusted reference | High; needs accurate database representation |
| Best sample type | Purified protein, gel band, enriched fraction | Complex or annotated proteomes |
| Primary output | Peptide tags, assembled sequence, coverage map | Protein IDs, peptide matches, modification sites |
| Speed and scale | Lower throughput, more analyst input | Higher throughput, better for large studies |
| Unknown sequence recovery | Strong | Weak when reference is absent |
| QC against expected design | Possible, but not the default use case | Strong when reference is reliable |
| Typical risk | Incomplete coverage, isobaric ambiguity | False match to wrong reference entry |
| Best follow-up | Terminal sequencing, intact mass, peptide mapping | Quantification, pathway analysis, PTM mapping |
This table shows why the methods are complementary. Database-based identification is optimized for scale and confirmation. De novo protein sequencing is optimized for sequence discovery and reference-independent evidence.
Which Approach Fits Different Study Goals
The best choice depends on what the project must prove.
Choose database-based identification when the sample comes from a well-annotated proteome, the expected sequence is represented in the database, and the goal is protein identification, quantification, or reference-backed confirmation. Examples include standard cell-line proteomics, pathway studies, and QC checks on proteins with reliable design records.
Choose de novo protein sequencing when the protein sequence is unknown, proprietary, engineered, truncated, or likely to differ from available references. Examples include unknown gel bands, legacy purified proteins, undocumented recombinant batches, and sequence recovery when genetic information is unavailable.
Choose a hybrid workflow when most peptides can be identified by database search, but a subset of high-value spectra require sequence derivation. This is common in antibody projects, biosimilar assessment, variant analysis, and biopharmaceutical comparability studies.
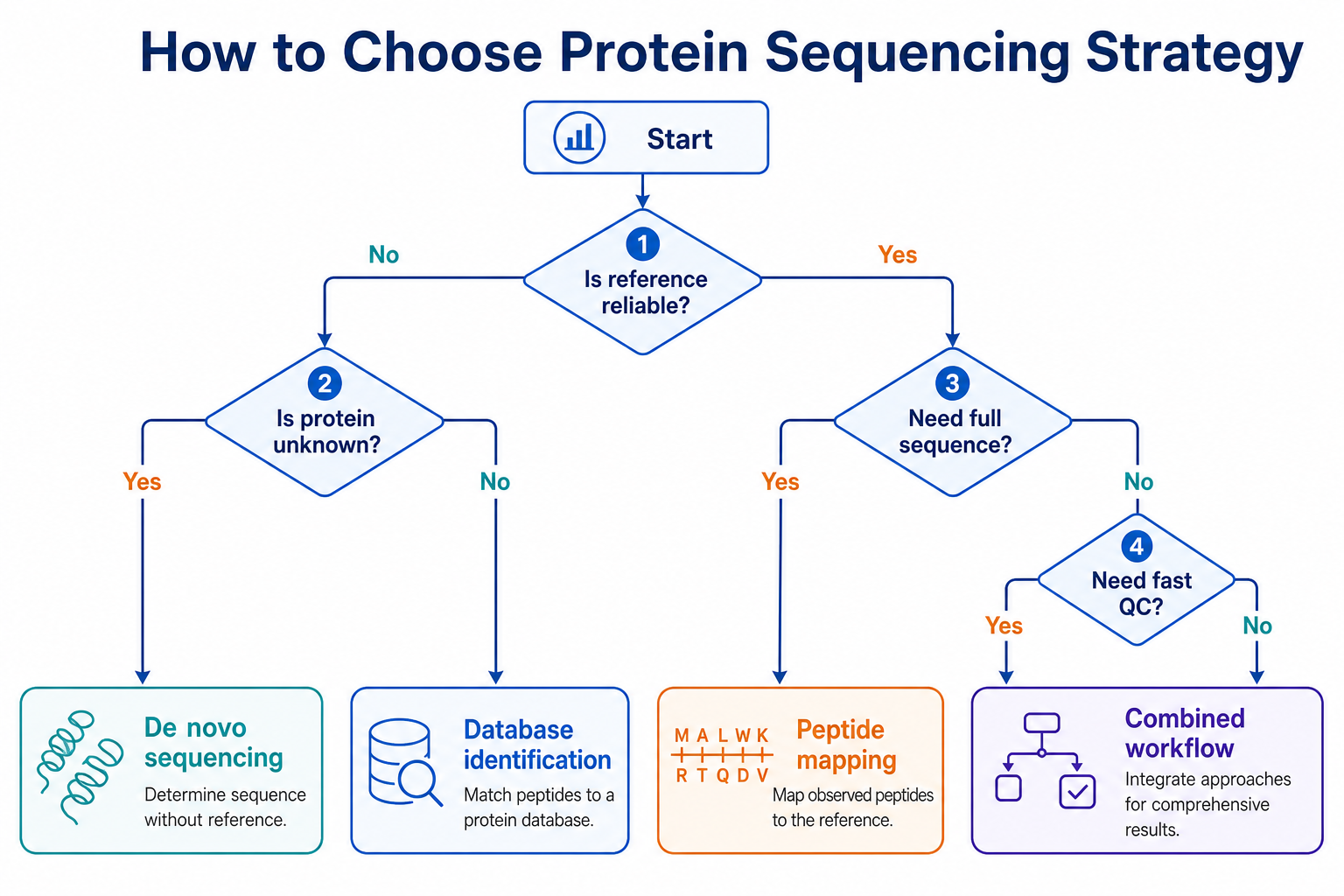
Figure 3. Method selection should follow reference reliability, protein novelty, coverage needs, and reporting urgency.
Decision Recommendations by Project Type
| Project Type | Recommended First Approach | Why |
|---|---|---|
| Standard proteomics on annotated samples | Database-based identification | Fast, scalable, statistically mature |
| Unknown protein band | De novo protein sequencing | Reference may not exist |
| Recombinant QC with trusted design | Database-based identification or peptide mapping | Expected sequence is known |
| Recombinant QC with sequence doubt | De novo protein sequencing | Variant or truncation may be present |
| Antibody variable-region recovery | De novo protein sequencing or hybrid workflow | Sequence divergence and complexity are common |
| Environmental or metaproteomics sample | Hybrid workflow | Mix of annotated and unannotated proteins |
| Publication-grade primary structure claim | De novo protein sequencing plus orthogonal confirmation | Higher evidence standard is required |
These recommendations are starting points. Sample purity, protein length, and modification status can shift the final plan.
Hybrid Workflows Often Provide the Best Balance
A strict either-or decision is not always necessary. A practical hybrid workflow may begin with database-based identification to characterize the majority of peptides, then apply de novo protein sequencing to unmatched spectra, low-scoring matches, or sequence-critical regions.
Hybrid strategies are useful when:
This approach preserves efficiency while still creating a path to novel or divergent sequence recovery.
Limitations to Keep in Mind
Database-based identification is only as good as the reference and search parameters. Incomplete databases, incorrect enzyme settings, and underestimated modification complexity can all reduce identification quality.
De novo protein sequencing depends on spectrum quality, enrichment, and expert assembly. It is not automatically superior simply because it does not use a database. Comparing the two methods only by number of identified peptides can lead to the wrong conclusion when the real need is sequence certainty.
Researchers should also define the deliverable before choosing. Protein names and relative abundance needs differ from residue-level sequence proof, cloning support, or regulatory documentation.
Practical Selection Checklist
Before starting the project, answer these questions:
If the answer to question 1 is yes and the goal is confirmation, database-based identification or peptide mapping is often sufficient. If the answer to question 2 is discovery, de novo protein sequencing should be planned from the start.
Frequently Asked Questions
1. Is de novo protein sequencing always better than database-based identification?
No. De novo protein sequencing is better when the reference is missing or unreliable. Database-based identification is usually better for large-scale identification and confirmation when the reference is accurate.
2. Can I use database search first and switch to de novo later?
Yes. A hybrid workflow is common. Database search can process most peptides efficiently, while de novo analysis targets unmatched or biologically important spectra.
3. Which method is better for recombinant protein QC?
If the intended sequence is trusted, database-based identification or peptide mapping is often enough. If the construct history is uncertain, de novo protein sequencing may be the safer route.
4. Which method works for unknown proteins?
De novo protein sequencing is the primary option when no suitable reference exists. Sample purity and enrichment still strongly affect the outcome.
5. How much evidence is needed for a strong sequence claim?
A strong claim usually requires overlapping peptides, replicate support, and clear documentation of unsupported regions. Terminal confirmation or intact mass measurement can strengthen the final report.
Conclusion
De novo protein sequencing and database-based identification answer different questions. Database-based identification is the efficient choice when the reference is reliable and the goal is identification or confirmation at scale. De novo protein sequencing is the stronger choice when the sequence is unknown, proprietary, variant-containing, or poorly represented in available databases. Many successful projects use a hybrid workflow that combines both approaches. The best decision starts with reference quality, sample type, and the level of sequence proof required. Researchers comparing these options for unknown proteins, recombinant materials, or antibody sequencing projects can contact MtoZ Biolabs to select a workflow aligned with the study goal and reporting standard.
How to order?

