





De Novo Protein Sequencing: Principles, Technical Challenges, and Research Applications
Introduction
Protein sequence is often treated as solved once a gene or construct sequence is available. In many research settings, that assumption breaks down. A purified band may not match any database entry. A recombinant product may differ from the intended construct. An antibody variable region may lack complete transcript data. A sample from a non-model organism may contain proteins with no reliable reference proteome. In these cases, researchers need sequence evidence derived from the protein itself rather than from an inferred database match.
This workflow addresses the need by assembling peptide-level MS/MS evidence into a longer protein sequence map. The method is built on peptide de novo interpretation, but the scientific value usually lies at the protein level: confirming a domain, identifying an unknown protein, validating a biologic, or documenting a sequence variant. Because protein-level assembly depends on overlapping peptides, spectrum quality, and manual review, the approach is more demanding than routine database-search proteomics.
Related Services
| Research Need | Recommended Service Direction |
|---|---|
| Confirm full protein sequence by mass spectrometry | |
| Derive peptide sequence without database matching | |
| Sequence unknown or poorly annotated proteins | |
| Sequence antibodies with incomplete reference data | |
| Support broader MS-based sequence analysis |
For projects that require protein-level sequence proof rather than peptide identification alone, MtoZ Biolabs can help evaluate whether protein-level sequencing, peptide mapping, antibody sequencing, or a combined MS workflow fits the sample and reporting goal.
What Is De Novo Protein Sequencing?
De novo protein sequencing is the process of determining protein sequence by assembling de novo-derived peptides into a contiguous or partially contiguous sequence map. Individual peptides are interpreted from MS/MS spectra without requiring a prior database match. Overlapping peptides from one or more protease digests are then aligned to build a longer sequence. The result may range from a confirmed local region to near full-length coverage, depending on sample quality, digestion design, and MS depth.
This approach differs from standard protein identification. Database search assigns spectra to known entries. De novo protein sequencing builds sequence evidence when the correct protein sequence is unknown, incomplete, proprietary, or potentially different from the expected construct. It also differs from Edman degradation, which reads N-terminal sequence stepwise. MS-based sequence analysis can analyze internal peptides from digested proteins and support broader coverage when multiple enzymes and overlapping peptides are used.
How Protein-Level Sequencing Works
A typical workflow begins with protein purification or enrichment. The protein is digested with one or more proteases to generate peptides suitable for LC-MS/MS analysis. High-quality MS/MS spectra are interpreted at the peptide level. Confirmed peptide sequences are then mapped onto a protein sequence scaffold using overlaps, shared residues, and complementary digest patterns.
Successful protein assembly depends on redundant coverage. A single peptide may confirm a short motif. Protein-level confidence increases when adjacent peptides overlap, when multiple enzymes produce consistent sequence segments, and when manual review resolves ambiguous residues. For antibodies, membrane proteins, and biopharmaceuticals, digestion strategy and fractionation are often customized to improve coverage of sequence-critical regions.
 | | Derive peptide sequence without database matching | [De Novo Sequencing Service](https://www.mtoz-biolabs.com/de-novo-sequencing.html) | | Sequence unknown or poorly annotated proteins | [Unknown Proteins Sequencing Service](https://www.mtoz-biolabs.com/sequencing-of-unknown-proteins.html) | | Sequence antibodies with incomplete reference data | [De Novo Antibody Sequencing Service](https://www.mtoz-biolabs.com/antibody-de-novo-sequencing.html) | | Support broader MS-based sequence analysis | [Protein Sequencing Service by Mass Spectrometry](https://www.mtoz-biolabs.com/sequence-analysis-based-on-mass-spectrometry.html) | For projects that require protein-level sequence proof rather than peptide identification alone, MtoZ Biolabs can help evaluate whether protein-level sequencing, peptide mapping, antibody sequencing, or a combined MS workflow fits the sample and reporting goal. ## What Is De Novo Protein Sequencing? De novo protein sequencing is the process of determining protein sequence by assembling de novo-derived peptides into a contiguous or partially contiguous sequence map. Individual peptides are interpreted from MS/MS spectra without requiring a prior database match. Overlapping peptides from one or more protease digests are then aligned to build a longer sequence. The result may range from a confirmed local region to near full-length coverage, depending on sample quality, digestion design, and MS depth. This approach differs from standard protein identification. Database search assigns spectra to known entries. De novo protein sequencing builds sequence evidence when the correct protein sequence is unknown, incomplete, proprietary, or potentially different from the expected construct. It also differs from Edman degradation, which reads N-terminal sequence stepwise. MS-based sequence analysis can analyze internal peptides from digested proteins and support broader coverage when multiple enzymes and overlapping peptides are used. ## How Protein-Level Sequencing Works A typical workflow begins with protein purification or enrichment. The protein is digested with one or more proteases to generate peptides suitable for LC-MS/MS analysis. High-quality MS/MS spectra are interpreted at the peptide level. Confirmed peptide sequences are then mapped onto a protein sequence scaffold using overlaps, shared residues, and complementary digest patterns. Successful protein assembly depends on redundant coverage. A single peptide may confirm a short motif. Protein-level confidence increases when adjacent peptides overlap, when multiple enzymes produce consistent sequence segments, and when manual review resolves ambiguous residues. For antibodies, membrane proteins, and biopharmaceuticals, digestion strategy and fractionation are often customized to improve coverage of sequence-critical regions.  <p align="center" alt=](https://file.mtoz-biolabs.com/pro/mtoz/20260701/2072247475358486528-de-novo-protein-sequencing-workflow.png) Figure 1. Protein-level sequencing workflow High-resolution MS/MS improves the ability to distinguish residues with similar mass and to interpret modified peptides. However, hardware alone does not complete the workflow. Expert interpretation remains important when spectra are sparse, when modifications shift fragment masses, or when only partial sequence coverage is achievable. The final report should distinguish high-confidence sequence segments from tentative or gap-containing regions. ## Sample Requirements Sample quality strongly influences whether protein-level assembly is feasible. Cleaner samples generally produce better spectra, fewer ambiguous assignments, and more efficient use of MS acquisition time. | Sample Factor | Recommended Condition | Why It Matters | | ---------------------- | ------------------------------------------------------------ | ------------------------------------------------------------ | | Sample format | Purified protein, enriched gel band, or prefractionated material | Reduces spectral complexity and improves target peptide recovery | | Purity | Single major band or highly enriched target | Lowers contaminant peptides that complicate assembly | | Protein amount | Sufficient for repeat digestion and replicate MS when possible | Low input limits overlap coverage and repeat analysis | | Buffer composition | Compatible with digestion; minimal interfering polymers or detergents | Harsh or complex buffers can reduce digestion efficiency | | Modifications | Disulfides, glycosylation, or blocked termini disclosed in advance | Modifications affect digestion, fragmentation, and interpretation | | Background information | Organism, expression system, expected size, or construct sequence if available | Helps scope digestion strategy and reporting depth | Gel-based samples should be excised cleanly with minimal keratin contamination. For antibody or recombinant protein projects, separating chains or domains before digestion can simplify interpretation. When sample amount is limited, project scope should be defined realistically. Partial coverage may still be valuable if the required region is captured with strong peptide evidence. ## Technical Challenges The method is powerful, but several technical challenges limit confidence if they are ignored. **Isoleucine and leucine ambiguity.** Many workflows cannot distinguish I and L by mass alone. Overlap with other sequence evidence or orthogonal methods may be needed for unambiguous assignment in critical regions. **Incomplete fragmentation.** Weak or sparse b-ion and y-ion series reduce confidence in peptide assembly. Long peptides, heavily modified residues, and poor precursor intensity all contribute to this problem. **Post-translational modifications.** Phosphorylation, glycosylation, oxidation, and other modifications can complicate de novo interpretation and protein overlap mapping. Modifications should be considered during digestion design and manual review. **Low protein abundance.** Low-abundance targets in complex backgrounds may require additional enrichment, fractionation, or targeted acquisition to obtain enough high-quality spectra for assembly. **Sequence assembly gaps.** Missing peptides, blocked termini, transmembrane regions, and repetitive sequence can leave gaps in the protein map. A gap does not always mean experimental failure, but gaps must be reported clearly. 
Figure 1. Protein-level sequencing workflow High-resolution MS/MS improves the ability to distinguish residues with similar mass and to interpret modified peptides. However, hardware alone does not complete the workflow. Expert interpretation remains important when spectra are sparse, when modifications shift fragment masses, or when only partial sequence coverage is achievable. The final report should distinguish high-confidence sequence segments from tentative or gap-containing regions. ## Sample Requirements Sample quality strongly influences whether protein-level assembly is feasible. Cleaner samples generally produce better spectra, fewer ambiguous assignments, and more efficient use of MS acquisition time. | Sample Factor | Recommended Condition | Why It Matters | | ---------------------- | ------------------------------------------------------------ | ------------------------------------------------------------ | | Sample format | Purified protein, enriched gel band, or prefractionated material | Reduces spectral complexity and improves target peptide recovery | | Purity | Single major band or highly enriched target | Lowers contaminant peptides that complicate assembly | | Protein amount | Sufficient for repeat digestion and replicate MS when possible | Low input limits overlap coverage and repeat analysis | | Buffer composition | Compatible with digestion; minimal interfering polymers or detergents | Harsh or complex buffers can reduce digestion efficiency | | Modifications | Disulfides, glycosylation, or blocked termini disclosed in advance | Modifications affect digestion, fragmentation, and interpretation | | Background information | Organism, expression system, expected size, or construct sequence if available | Helps scope digestion strategy and reporting depth | Gel-based samples should be excised cleanly with minimal keratin contamination. For antibody or recombinant protein projects, separating chains or domains before digestion can simplify interpretation. When sample amount is limited, project scope should be defined realistically. Partial coverage may still be valuable if the required region is captured with strong peptide evidence. ## Technical Challenges The method is powerful, but several technical challenges limit confidence if they are ignored. **Isoleucine and leucine ambiguity.** Many workflows cannot distinguish I and L by mass alone. Overlap with other sequence evidence or orthogonal methods may be needed for unambiguous assignment in critical regions. **Incomplete fragmentation.** Weak or sparse b-ion and y-ion series reduce confidence in peptide assembly. Long peptides, heavily modified residues, and poor precursor intensity all contribute to this problem. **Post-translational modifications.** Phosphorylation, glycosylation, oxidation, and other modifications can complicate de novo interpretation and protein overlap mapping. Modifications should be considered during digestion design and manual review. **Low protein abundance.** Low-abundance targets in complex backgrounds may require additional enrichment, fractionation, or targeted acquisition to obtain enough high-quality spectra for assembly. **Sequence assembly gaps.** Missing peptides, blocked termini, transmembrane regions, and repetitive sequence can leave gaps in the protein map. A gap does not always mean experimental failure, but gaps must be reported clearly.  Figure 2. Common technical challenges in protein-level sequencing
These challenges explain why protein-level reporting should include confidence labeling. A sequence map without gap annotation or ambiguous residue flags can create a false sense of completeness. Transparent reporting is especially important in antibody sequencing, biosimilar assessment, and unknown protein identification. ## Expected Outputs and Reporting Depth Reporting depth should match the biological or commercial decision behind the project. Not every study requires full-length sequence coverage, but every study should define what level of evidence is sufficient before analysis begins. | Output Type | Typical Content | Best Used For | | -------------------------- | ------------------------------------------------------ | ------------------------------------------------------- | | Confirmed peptide list | De novo-derived peptides with spectrum support | Regional confirmation and exploratory mapping | | Protein sequence map | Overlapping peptides assembled into a longer sequence | Clone design, homology analysis, construct verification | | Annotated spectra | Key MS/MS spectra linked to peptide assignments | Manual review, publication, or audit support | | Ambiguity flags | I/L positions, low-confidence residues, or gap regions | Transparent reporting and follow-up validation | | Coverage summary | Percent or region-based coverage of the target protein | QC, comparability, and project acceptance criteria | | Validation recommendations | Suggested orthogonal checks for critical regions | Antibody, biologic, or high-stakes sequence claims | A strong sequence report separates high-confidence segments from tentative calls. It should also indicate where additional digestion, deeper MS, or orthogonal validation would most improve the final sequence evidence. ## Research Applications The method is used when protein sequence itself is the scientific deliverable. **Unknown protein identification.** A purified protein with no reliable database match can be analyzed to generate sequence candidates for cloning, functional study, or homology searching. **Antibody sequence confirmation.** Variable-region sequence may be required when hybridoma transcript data are incomplete or when expressed antibody products must be verified at the protein level. **Recombinant protein quality control.** Expressed proteins can be checked for sequence accuracy, truncations, substitutions, or expression-related sequence drift. **Variant and mutation analysis.** Unexpected sequence differences from a reference construct can be detected when variant-containing peptides are recovered and assembled. **Non-model organism proteomics.** Proteins from poorly annotated species may yield informative sequence segments even when full genomic context is limited. Figure 3. Research applications of the method
In each application, the required coverage level should be defined early. A short confirmed region may be enough for one project, while another may require maximal overlap across a full chain or domain. ## Advantages and Current Limits The main advantage is direct protein-level sequence evidence when references are missing or unreliable. It supports discovery, engineering validation, and documentation in settings where DNA sequence alone is insufficient. Current limits are equally important. Full-length coverage is not guaranteed for every sample. Manual interpretation adds time and expertise. Highly complex mixtures, poor digestion, and repetitive protein regions can reduce the amount of usable sequence. The workflow is therefore most effective when combined with realistic project scoping, strong sample preparation, and clear reporting standards. ## Future Outlook MS-based sequence workflows are likely to continue integrating database search, de novo peptide analysis, targeted MS, chemical derivatization, and orthogonal methods such as Edman degradation or gene sequencing. Improvements in mass accuracy, acquisition speed, and interpretation software will help, but protein-level assembly will still depend on overlap design and expert review for high-confidence reporting. The most useful future model is not full automation of protein sequence proof. It is a layered workflow in which protein-level sequencing fills the gaps that reference-based methods cannot cover, while validation and reporting standards keep pace with the biological decision being made. ## Frequently Asked Questions ### 1. What is the difference between de novo sequencing and de novo protein sequencing? De novo sequencing usually refers to peptide sequence interpretation from MS/MS spectra. De novo protein sequencing extends that process by assembling multiple peptides into a longer protein sequence map. ### 2. Can this method provide full-length sequence? Sometimes. Full-length coverage depends on sample purity, digestion strategy, MS depth, protein properties, and the presence of blocked termini or difficult regions. Many projects achieve strong regional or domain coverage even when full-length assembly is incomplete. ### 3. What sample types are most suitable? Purified proteins, enriched gel bands, antibody chains, and recombinant products are commonly suitable. Complex crude lysates are more difficult unless the target is strongly enriched. ### 4. How is this approach different from database search? Database search matches spectra to known sequences. The method builds sequence evidence when the correct protein sequence is not available or may differ from the reference. ### 5. When should researchers choose protein-level sequencing? Choose this approach when protein sequence proof is required and database-based identification is insufficient, especially for unknown proteins, engineered biologics, antibody products, and variant-containing samples. ## Conclusion De novo protein sequencing provides a practical route to protein-level sequence evidence when references are incomplete or untrustworthy. The workflow depends on high-quality samples, thoughtful digestion design, strong MS/MS data, overlap assembly, and transparent reporting of gaps and ambiguous residues. It is widely used for unknown protein identification, antibody confirmation, recombinant protein QC, variant analysis, and non-model organism research. For projects that need dependable protein sequence reporting beyond database matching, contact MtoZ Biolabs to discuss protein-level sequencing, unknown protein analysis, antibody sequencing, or an integrated MS-based sequence workflow.">Figure 1. Protein-level sequencing workflow
High-resolution MS/MS improves the ability to distinguish residues with similar mass and to interpret modified peptides. However, hardware alone does not complete the workflow. Expert interpretation remains important when spectra are sparse, when modifications shift fragment masses, or when only partial sequence coverage is achievable. The final report should distinguish high-confidence sequence segments from tentative or gap-containing regions.
Sample Requirements
Sample quality strongly influences whether protein-level assembly is feasible. Cleaner samples generally produce better spectra, fewer ambiguous assignments, and more efficient use of MS acquisition time.
| Sample Factor | Recommended Condition | Why It Matters |
|---|---|---|
| Sample format | Purified protein, enriched gel band, or prefractionated material | Reduces spectral complexity and improves target peptide recovery |
| Purity | Single major band or highly enriched target | Lowers contaminant peptides that complicate assembly |
| Protein amount | Sufficient for repeat digestion and replicate MS when possible | Low input limits overlap coverage and repeat analysis |
| Buffer composition | Compatible with digestion; minimal interfering polymers or detergents | Harsh or complex buffers can reduce digestion efficiency |
| Modifications | Disulfides, glycosylation, or blocked termini disclosed in advance | Modifications affect digestion, fragmentation, and interpretation |
| Background information | Organism, expression system, expected size, or construct sequence if available | Helps scope digestion strategy and reporting depth |
Gel-based samples should be excised cleanly with minimal keratin contamination. For antibody or recombinant protein projects, separating chains or domains before digestion can simplify interpretation. When sample amount is limited, project scope should be defined realistically. Partial coverage may still be valuable if the required region is captured with strong peptide evidence.
Technical Challenges
The method is powerful, but several technical challenges limit confidence if they are ignored.
Isoleucine and leucine ambiguity. Many workflows cannot distinguish I and L by mass alone. Overlap with other sequence evidence or orthogonal methods may be needed for unambiguous assignment in critical regions.
Incomplete fragmentation. Weak or sparse b-ion and y-ion series reduce confidence in peptide assembly. Long peptides, heavily modified residues, and poor precursor intensity all contribute to this problem.
Post-translational modifications. Phosphorylation, glycosylation, oxidation, and other modifications can complicate de novo interpretation and protein overlap mapping. Modifications should be considered during digestion design and manual review.
Low protein abundance. Low-abundance targets in complex backgrounds may require additional enrichment, fractionation, or targeted acquisition to obtain enough high-quality spectra for assembly.
Sequence assembly gaps. Missing peptides, blocked termini, transmembrane regions, and repetitive sequence can leave gaps in the protein map. A gap does not always mean experimental failure, but gaps must be reported clearly.
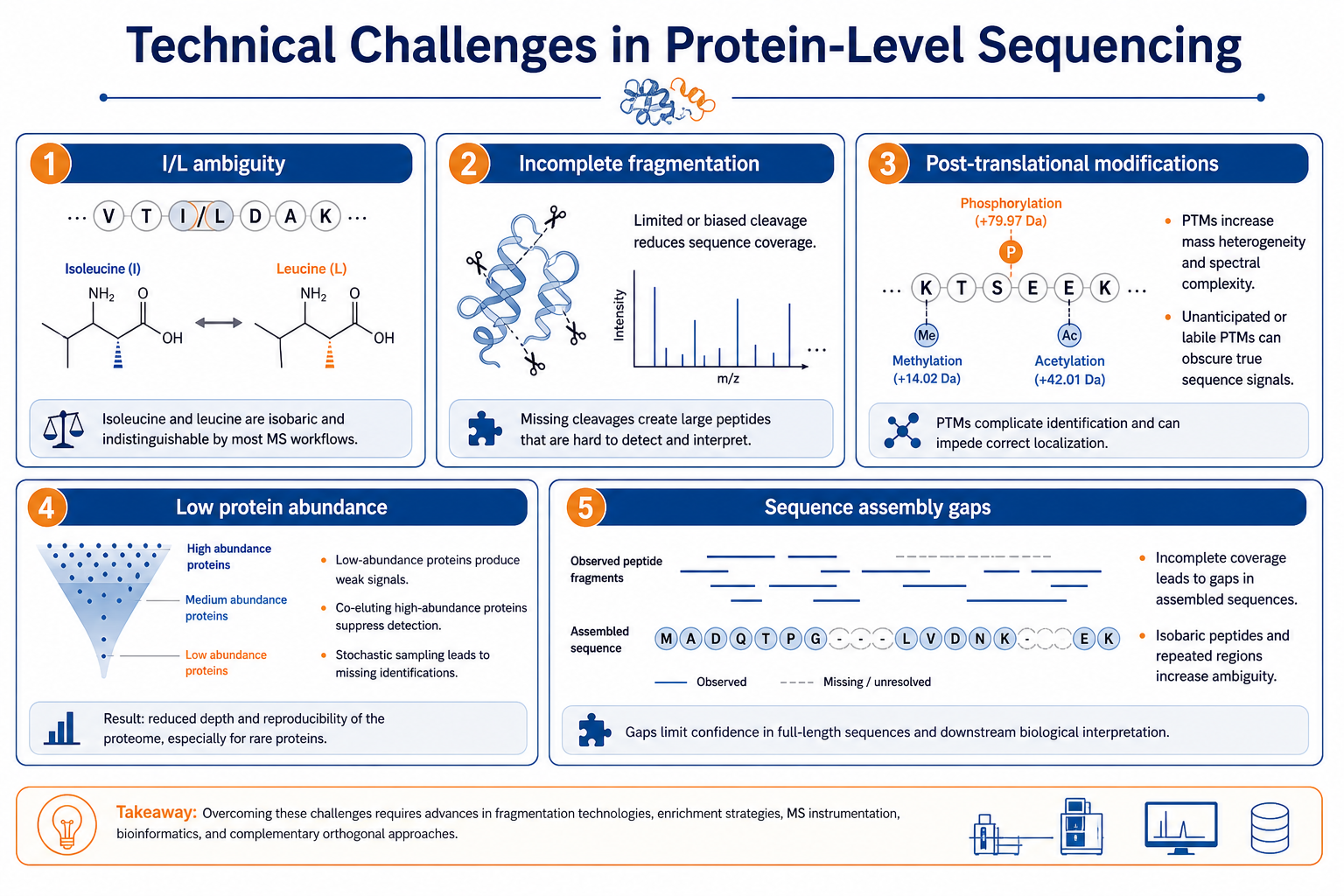
Figure 2. Common technical challenges in protein-level sequencing
These challenges explain why protein-level reporting should include confidence labeling. A sequence map without gap annotation or ambiguous residue flags can create a false sense of completeness. Transparent reporting is especially important in antibody sequencing, biosimilar assessment, and unknown protein identification.
Expected Outputs and Reporting Depth
Reporting depth should match the biological or commercial decision behind the project. Not every study requires full-length sequence coverage, but every study should define what level of evidence is sufficient before analysis begins.
| Output Type | Typical Content | Best Used For |
|---|---|---|
| Confirmed peptide list | De novo-derived peptides with spectrum support | Regional confirmation and exploratory mapping |
| Protein sequence map | Overlapping peptides assembled into a longer sequence | Clone design, homology analysis, construct verification |
| Annotated spectra | Key MS/MS spectra linked to peptide assignments | Manual review, publication, or audit support |
| Ambiguity flags | I/L positions, low-confidence residues, or gap regions | Transparent reporting and follow-up validation |
| Coverage summary | Percent or region-based coverage of the target protein | QC, comparability, and project acceptance criteria |
| Validation recommendations | Suggested orthogonal checks for critical regions | Antibody, biologic, or high-stakes sequence claims |
A strong sequence report separates high-confidence segments from tentative calls. It should also indicate where additional digestion, deeper MS, or orthogonal validation would most improve the final sequence evidence.
Research Applications
The method is used when protein sequence itself is the scientific deliverable.
Unknown protein identification. A purified protein with no reliable database match can be analyzed to generate sequence candidates for cloning, functional study, or homology searching.
Antibody sequence confirmation. Variable-region sequence may be required when hybridoma transcript data are incomplete or when expressed antibody products must be verified at the protein level.
Recombinant protein quality control. Expressed proteins can be checked for sequence accuracy, truncations, substitutions, or expression-related sequence drift.
Variant and mutation analysis. Unexpected sequence differences from a reference construct can be detected when variant-containing peptides are recovered and assembled.
Non-model organism proteomics. Proteins from poorly annotated species may yield informative sequence segments even when full genomic context is limited.
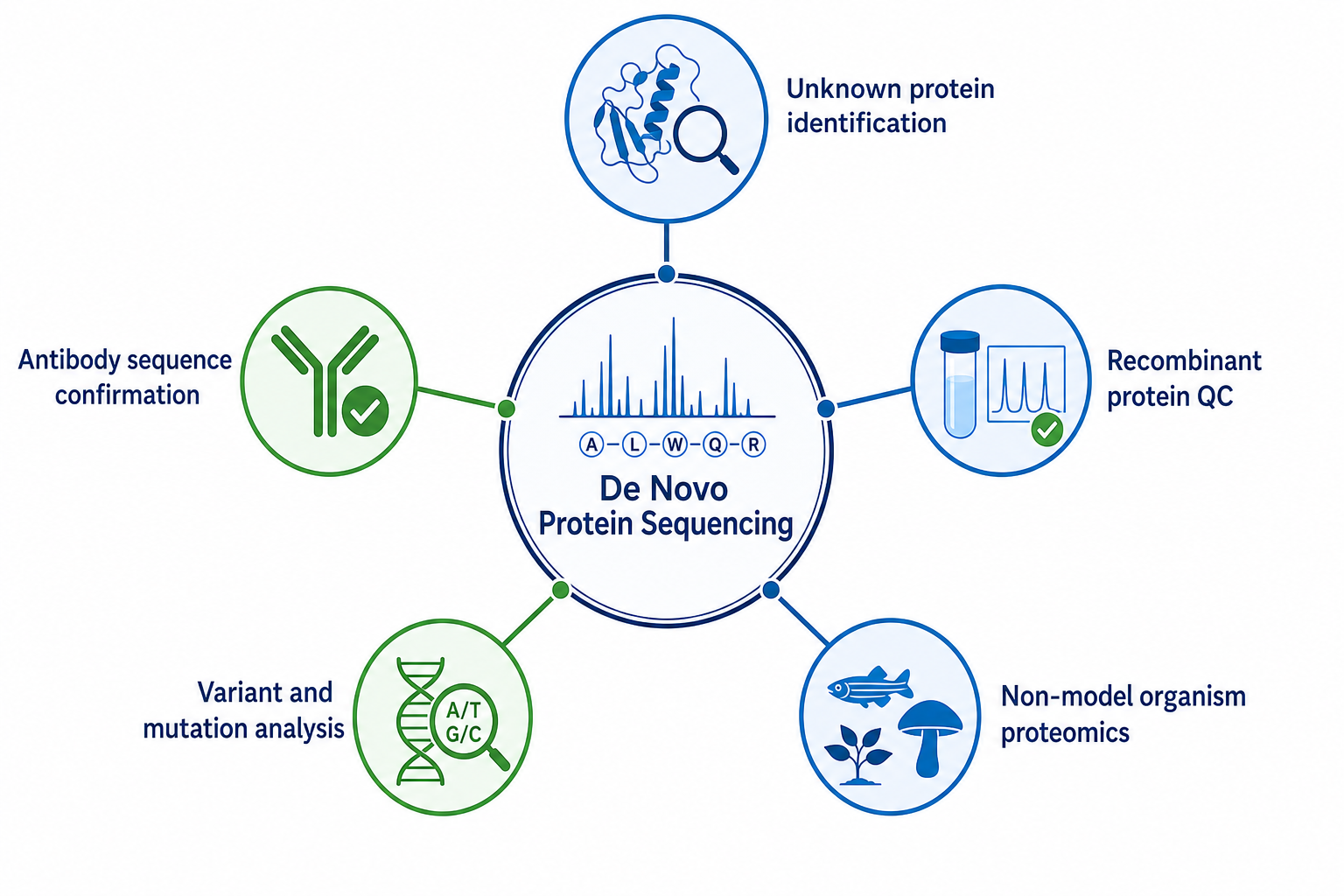 Figure 3. Research applications of the method
Figure 3. Research applications of the method
In each application, the required coverage level should be defined early. A short confirmed region may be enough for one project, while another may require maximal overlap across a full chain or domain.
Advantages and Current Limits
The main advantage is direct protein-level sequence evidence when references are missing or unreliable. It supports discovery, engineering validation, and documentation in settings where DNA sequence alone is insufficient.
Current limits are equally important. Full-length coverage is not guaranteed for every sample. Manual interpretation adds time and expertise. Highly complex mixtures, poor digestion, and repetitive protein regions can reduce the amount of usable sequence. The workflow is therefore most effective when combined with realistic project scoping, strong sample preparation, and clear reporting standards.
Future Outlook
MS-based sequence workflows are likely to continue integrating database search, de novo peptide analysis, targeted MS, chemical derivatization, and orthogonal methods such as Edman degradation or gene sequencing. Improvements in mass accuracy, acquisition speed, and interpretation software will help, but protein-level assembly will still depend on overlap design and expert review for high-confidence reporting.
The most useful future model is not full automation of protein sequence proof. It is a layered workflow in which protein-level sequencing fills the gaps that reference-based methods cannot cover, while validation and reporting standards keep pace with the biological decision being made.
Frequently Asked Questions
1. What is the difference between de novo sequencing and de novo protein sequencing?
De novo sequencing usually refers to peptide sequence interpretation from MS/MS spectra. De novo protein sequencing extends that process by assembling multiple peptides into a longer protein sequence map.
2. Can this method provide full-length sequence?
Sometimes. Full-length coverage depends on sample purity, digestion strategy, MS depth, protein properties, and the presence of blocked termini or difficult regions. Many projects achieve strong regional or domain coverage even when full-length assembly is incomplete.
3. What sample types are most suitable?
Purified proteins, enriched gel bands, antibody chains, and recombinant products are commonly suitable. Complex crude lysates are more difficult unless the target is strongly enriched.
4. How is this approach different from database search?
Database search matches spectra to known sequences. The method builds sequence evidence when the correct protein sequence is not available or may differ from the reference.
5. When should researchers choose protein-level sequencing?
Choose this approach when protein sequence proof is required and database-based identification is insufficient, especially for unknown proteins, engineered biologics, antibody products, and variant-containing samples.
Conclusion
De novo protein sequencing provides a practical route to protein-level sequence evidence when references are incomplete or untrustworthy. The workflow depends on high-quality samples, thoughtful digestion design, strong MS/MS data, overlap assembly, and transparent reporting of gaps and ambiguous residues. It is widely used for unknown protein identification, antibody confirmation, recombinant protein QC, variant analysis, and non-model organism research.
For projects that need dependable protein sequence reporting beyond database matching, contact MtoZ Biolabs to discuss protein-level sequencing, unknown protein analysis, antibody sequencing, or an integrated MS-based sequence workflow.
How to order?

