





Common PhIP-Seq Failure Points: Low Enrichment, High Background, and Replicate Mismatch
- Does the peptide remain enriched after control-aware filtering?
- Do neighboring peptides support a plausible linear epitope region?
- Does the pattern persist across replicates?
- Reanalyze first when sequencing and controls are broadly usable but hit confidence depends too much on threshold settings.
- Repeat immunoprecipitation first when sequencing is acceptable but enrichment or replicate stability is still weak.
- Resequence first when mapped depth or library representation limits interpretation.
- Move to orthogonal validation or redesign when conclusions rest on isolated peptides, weak adjacency support, or unstable replicates.
Low enrichment, high background, and replicate mismatch in PhIP-Seq usually point to different next moves. Weak recovery of expected positive controls usually points back to immunoprecipitation performance. Strong peptide retention in mock IP or bead-only controls usually points to non-specific binding and background modeling. Replicates that split mainly near the count floor often reflect unstable ranking in sparse data, not immediate proof that the biology is absent.
That distinction matters. Post-run troubleshooting is not about rescuing every weak dataset with harder thresholds. The practical question is whether the data can be clarified through reanalysis, whether a wet-lab repeat is warranted, or whether the project should move to orthogonal confirmation.
Where Failure Patterns Usually Appear
A PhIP-Seq run can look fine at the FASTQ and mapping level and still be hard to trust. Teams usually run into one of three patterns. First, expected reactive peptides show only modest separation from input or controls. Second, negative controls retain long peptide lists that overlap with apparent hits. Third, biological or technical replicates produce different top-ranked peptide sets even when total read depth looks acceptable.
For troubleshooting teams and sequencing analysts, these patterns create a real decision bottleneck. A translational lab may need to know whether a candidate antigen is worth follow-up. A bioinformatics group may need to tell weak biology from assay noise. A project lead may need to decide whether remaining sample should go to another immunoprecipitation round, a resequencing run, or a targeted validation assay.
The Four Cause Categories Worth Checking First
Most unstable PhIP-Seq output falls into four categories. Keeping the review this narrow helps because broad, full-workflow checklists rarely change the next decision.
1. Under-enrichment during immunoprecipitation
If positive controls recover poorly and IP samples stay close to the input distribution, under-enrichment is the first suspect. Capture efficiency may be low, wash conditions may be too aggressive for weak true binders, or bead handling may vary across wells. In this pattern, sequencing can be technically usable while biological separation stays weak.
2. Persistent background in controls
High background usually shows up as recurrent peptide retention in mock IP, bead-only controls, or unrelated negative controls. Some phage-displayed peptides bind beads or matrix components more readily than others, and those recurrent peptides can take over the hit list if control-aware filtering is too loose.
3. Sparse-count instability
Replicate mismatch often starts with peptides near the detection floor. Small differences in sampling, normalization, or shrinkage can shift rank order enough to change the reported hit set. That is different from strong-signal disagreement, where the same well-supported peptide behaves inconsistently across replicates.
4. Uneven library representation or sequencing composition
Low mapped depth, barcode imbalance, regional library dropout, or underrepresented peptide subsets can flatten real enrichment. Missing signal is sometimes a coverage problem rather than a true lack of antibody binding, especially when affected peptides cluster in specific regions of the displayed library.
Related Services
Main Service |
Supporting Service |
Validation Service |
Alternative Service |
Step-by-Step Troubleshooting Workflow
Step 1: Classify the dominant failure signature
Review four outputs together: mapped reads, control behavior, positive control recovery, and replicate concordance. More than one issue can appear at once, but one usually drives the next decision. This step should end with a clear classification supported by the count matrix and controls.
Step 2: Confirm that sequencing output can support interpretation
Before repeating the assay, inspect usable mapped reads, library member coverage, barcode balance, and count distribution across the library. If coverage is uneven or dropout is obvious, another immunoprecipitation round will not fix the main problem. Resequencing or library review is the better first move.
A good checkpoint is whether the matrix supports stable enrichment scoring. If small normalization changes reshuffle the candidate list, the dataset may be too sparse or uneven for confident peptide-level interpretation.
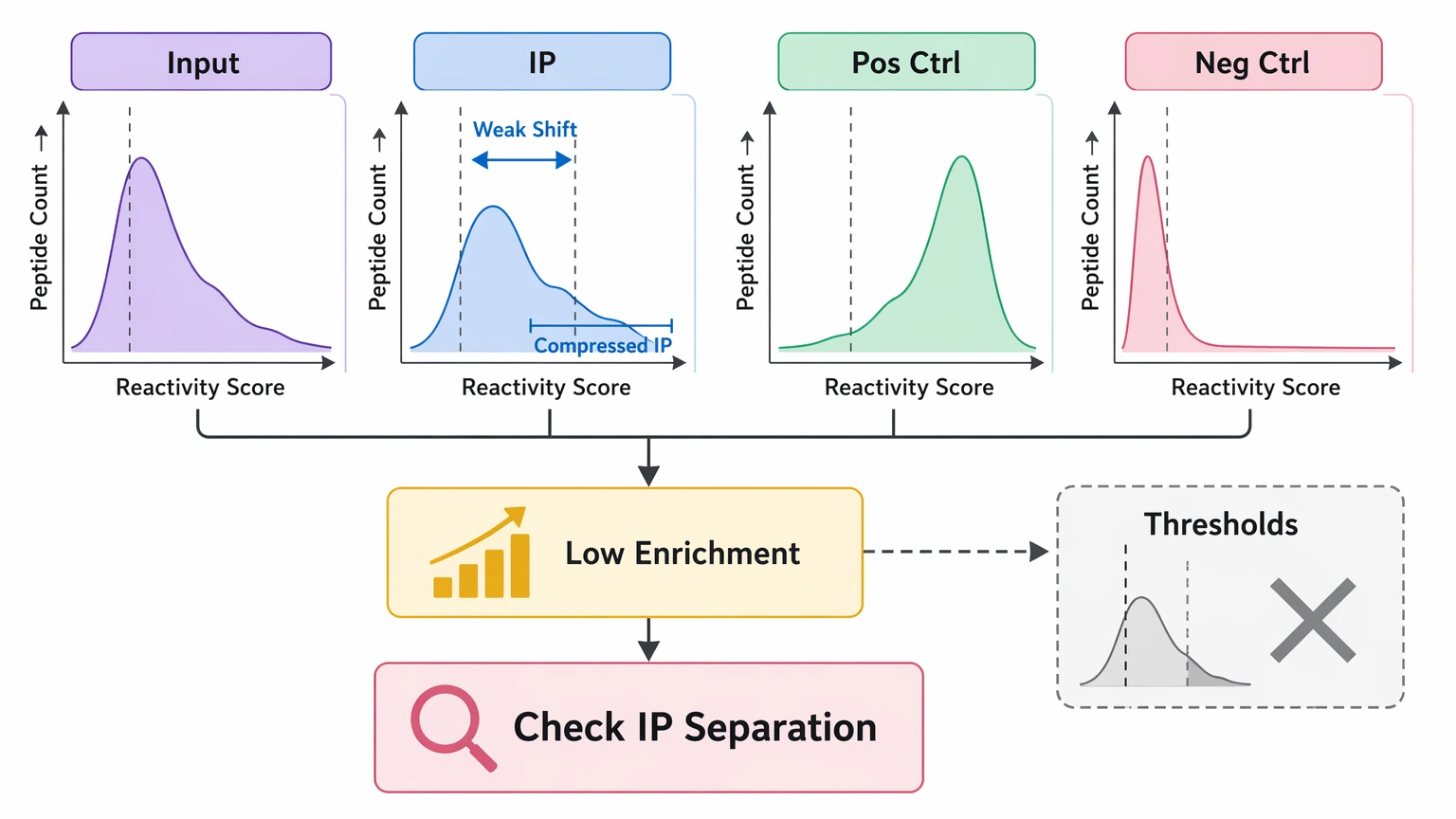
Step 3: If low enrichment dominates, evaluate IP separation rather than tightening thresholds
When controls are fairly clean but enrichment is weak, compare input, IP, positive control, and negative control profiles directly. Look for a real shift in expected reactive peptides after immunoprecipitation. If the full IP distribution stays compressed relative to input, stricter thresholds rarely help. They usually remove borderline candidates without improving specificity.
A selective repeat of immunoprecipitation is often the better next step, especially with tighter handling control and closer monitoring of positive controls. If your team needs to decide whether weak separation reflects assay execution or limited biology, contact MtoZ Biolabs to submit your requirements and review the count matrix, control behavior, and replicate structure before spending more sample.
Step 4: If high background dominates, rebuild hit confidence around control-aware filtering
When negative controls retain many peptides, treat those controls as core evidence, not side information. Identify peptides that recur across mock IPs, bead-only controls, batches, or lots. Those recurrent peptides should be downgraded unless sample signal rises above control behavior by a convincing margin.
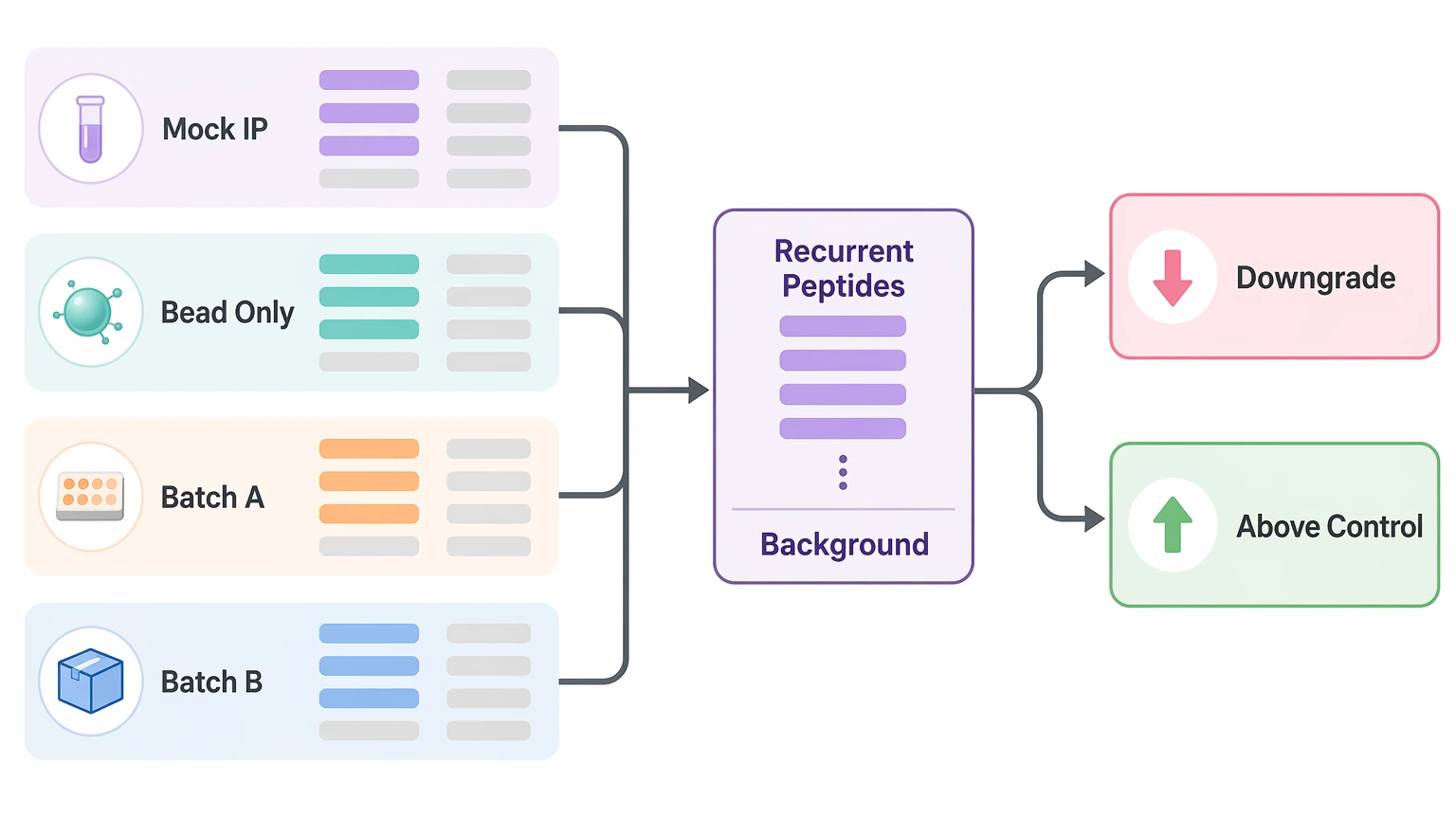
Then review each candidate hit with three practical questions:
A short hit list that survives those checks is more useful than a long list driven by sticky peptides.
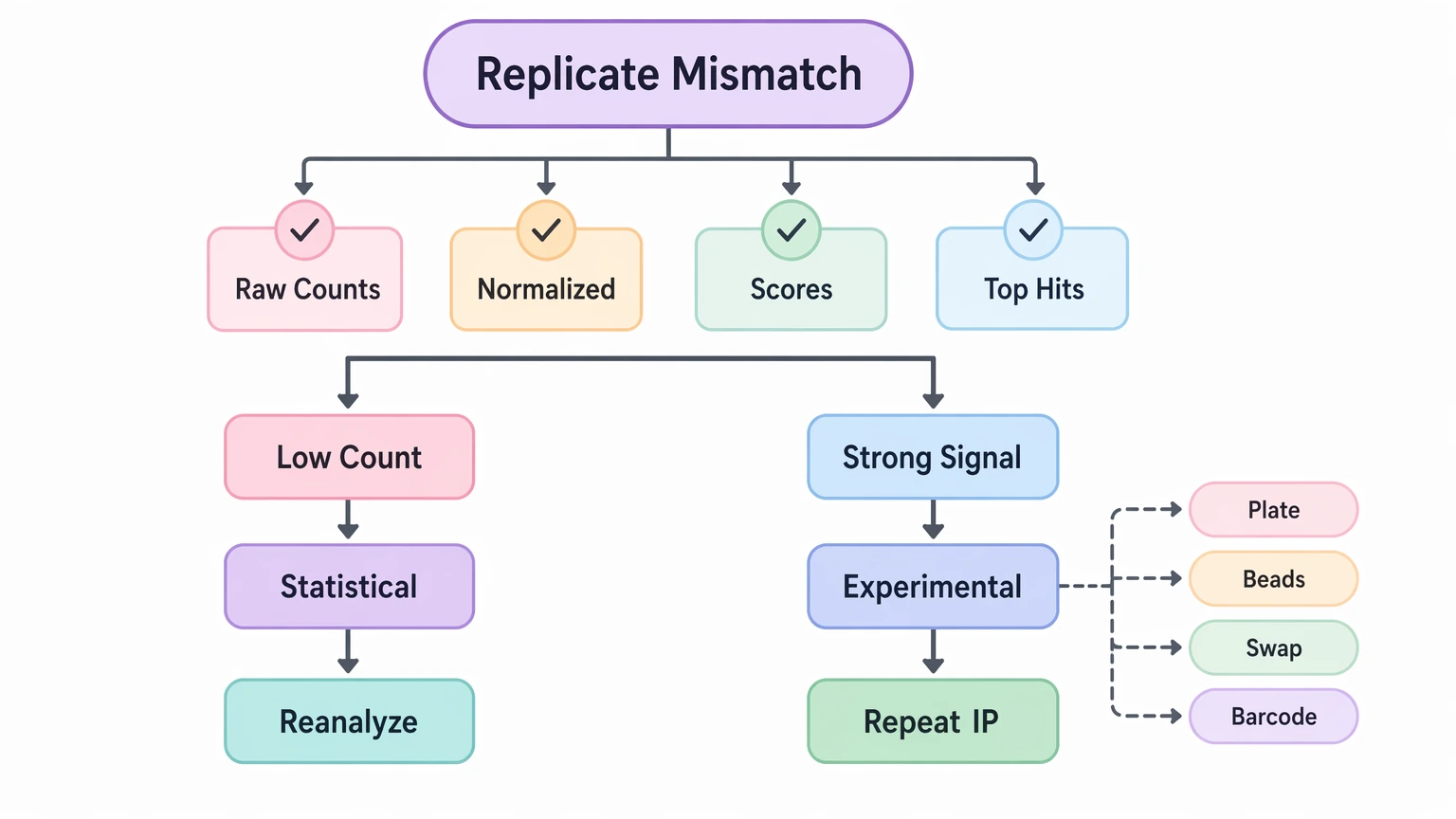
Step 5: If replicate mismatch dominates, separate statistical instability from assay instability
Check concordance at more than one level: raw counts, normalized counts, enrichment scores, and overlap among top-ranked peptides. If disagreement is concentrated in low-count peptides with marginal enrichment, the issue is often statistical. Reanalysis with minimum count support, steadier normalization, and clearer confidence grading is usually appropriate before any wet-lab repeat.
If mismatch extends to stronger signals, positive controls, or multiple neighboring peptides from the same region, suspect experimental instability instead. Plate effects, bead handling variation, sample swaps, or barcode assignment problems fit that pattern better. That result supports repeating immunoprecipitation or revising replicate handling rather than continuing to tune the analysis.
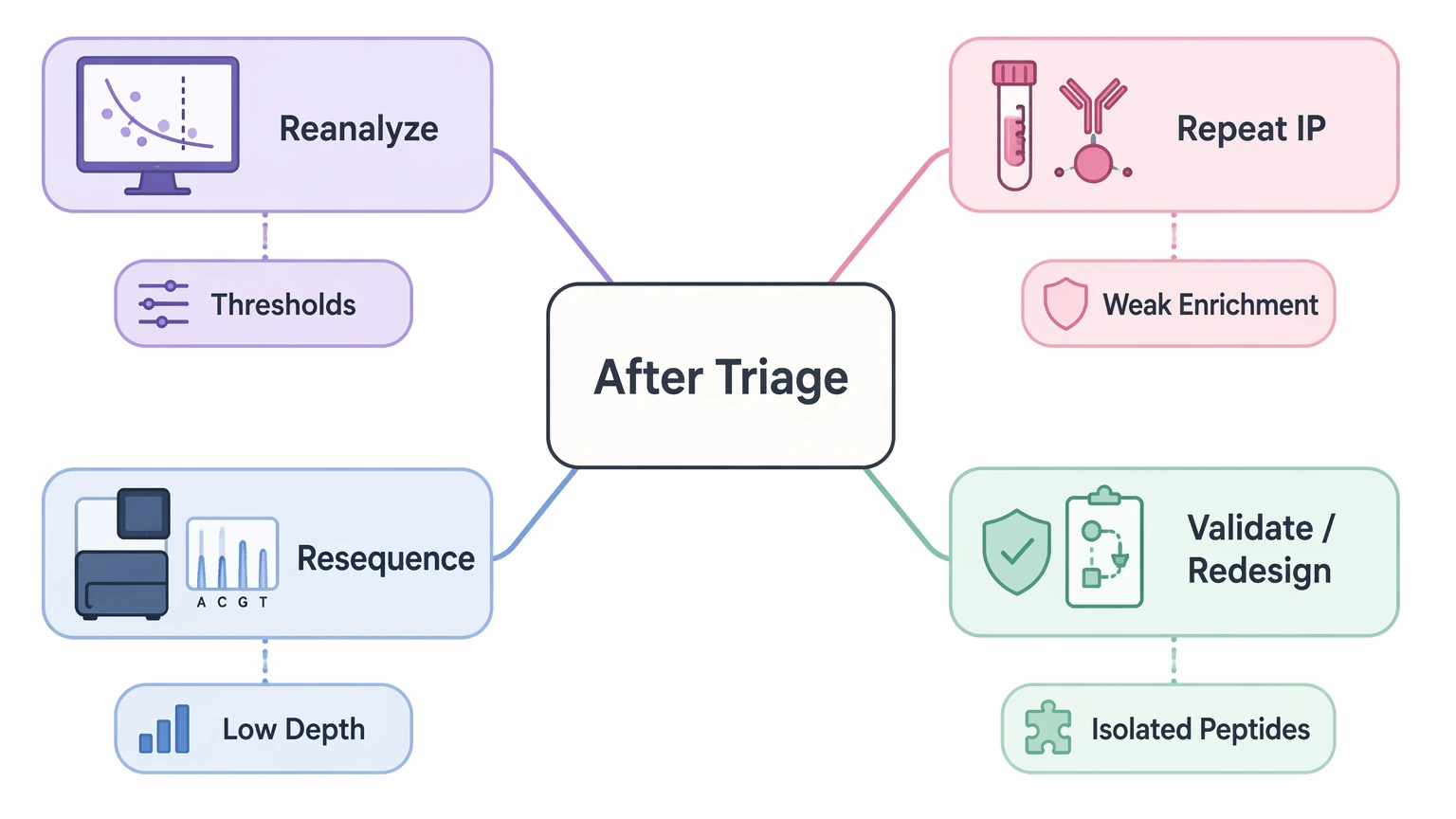
Step 6: Choose the next action path
After triage, most projects fit one of four paths:
If your group is deciding between rerun planning and confirmation testing, MtoZ Biolabs can evaluate your project if you contact us with the count matrix, replicate map, control definitions, and the specific decision that has to be made next.
Expected Results and Validation Methods
A successful troubleshooting pass should produce three practical outputs: a narrowed failure category, a revised hit list with the affected control or replicate rule documented, and a validation plan that matches the remaining confidence level. Low-enrichment cases usually need IP separation review before validation. High-background cases need control-aware filtering and recurrent-peptide review. Replicate-mismatch cases need a decision about whether the pattern reflects sparse-count instability, batch behavior, or a biological subgroup.
Validation should be matched to the corrected failure mode rather than applied as a generic final step. Candidate linear epitopes can move to peptide ELISA or peptide array follow-up. Antigen-level questions may need protein array, immunoblot, or antigen immunoprecipitation depending on format and sample availability. If the study cannot preserve enough material for confirmation, the report should label the result as exploratory rather than validation-ready.
Key Cautions and Practical Constraints
Do not treat deeper sequencing as a universal fix for weak enrichment. More reads cannot rescue poor IP separation, missing controls, or library representation problems that were already present before sequencing. Do not tighten thresholds until the dominant failure mode is known, because stricter filters can remove true but sparse peptide patterns while leaving recurrent background intact.
PhIP-Seq troubleshooting also depends on sample matrix, cohort design, and available controls. Serum, plasma, and CSF can produce different background behavior, and small cohorts may not support stable empirical null models. When failure patterns affect downstream assay choice, MtoZ Biolabs can review the sample context, control plan, and validation reserve before the next run is designed.
Interpretation Guardrails That Prevent Overcalling
A peptide-level hit is not automatically a protein-level conclusion. Confidence rises when enrichment is backed by clean control separation, replicate agreement, and adjacent peptide coverage across a coherent region. Confidence drops when the signal rests on a single isolated peptide, weak fold change, or unstable ranking across analysis settings.
It also helps to keep the assay boundary clear. PhIP-Seq is strongest for linear epitope-scale antibody profiling. It does not reliably capture every conformational interaction, and it should not be treated as a calibrated measure of absolute antibody concentration. A weak or missing peptide signal may reflect assay limits, incomplete representation, or genuinely low reactivity, and those possibilities should be weighed before a project is advanced or stopped.
Conclusion
PhIP-Seq troubleshooting works best when the dominant failure signature drives the workflow. Low enrichment usually points to immunoprecipitation performance, high background calls for stricter control-aware interpretation, and replicate mismatch requires a deliberate split between sparse-count noise and true assay instability. For antibody profiling projects with weak signal, noisy controls, or uncertain hit confidence, a structured review of controls, mapped reads, normalization stability, and adjacent peptide coverage usually shows whether the dataset is analytically salvageable, better suited to a selective rerun, or better advanced through orthogonal validation. If your project is at that point, prepare the count matrix, control summary, and replicate layout, then seek a technical review to evaluate the project and choose the most defensible next step.
FAQ
When should mock IP controls be considered batch-specific rather than global?
Use batch-specific controls when bead lots, operators, wash conditions, or plate layouts changed across runs. A global background list can miss local artifacts or over-filter a batch that was unusually clean.
Is deeper sequencing always the right response to weak signal?
No. More reads help only when inadequate depth or uneven representation is the real bottleneck. If positive controls already fail to separate after immunoprecipitation, deeper sequencing mostly adds counts from a weakly enriched pool.
How many adjacent peptides are enough to support a linear epitope interpretation?
There is no universal cutoff, but confidence improves when enrichment extends across overlapping peptides in one contiguous region rather than appearing as a single isolated spike. The exact pattern depends on library tiling density and peptide design.
Can normalization alone create apparent replicate disagreement?
Yes. When many peptides sit close to background, different normalization or shrinkage strategies can move borderline candidates above or below the reporting threshold. That is why replicate review should include both raw and normalized views.
When is protein array follow-up more informative than another PhIP-Seq repeat?
A protein array can help when the main question is broader antigen-level screening or when conformational binding is a serious concern. It is not a direct substitute for every peptide-level interpretation question.
What materials make an external troubleshooting review efficient?
A compact package is usually enough: count matrix, sample sheet, replicate map, control definitions, mapped-read summary, library reference, normalization method, and the short list of biological questions the dataset must answer.
How to order?

