





Antibody Protein Sequencing: How Mass Spectrometry Reconstructs Antibody Variable-Region Sequences
- heavy-chain variable region (VH)
- light-chain variable region (VL)
- CDR1, CDR2, and CDR3 boundaries where coverage supports annotation
- framework region assignments
- documented gaps, ambiguous residues, or low-confidence segments
- confirming sample purity before digestion
- checking that VH and VL lengths and framework patterns are plausible
- comparing independent digests or technical replicates when possible
- using orthogonal confirmation for critical rescue or expression decisions
- documenting ambiguous residues or unsupported CDR segments clearly
Introduction
Antibody variable-region sequences define antigen recognition, yet many projects still lack a reliable genetic record for the antibody in hand. Hybridoma cells may be lost, expression plasmids may be incomplete, and legacy purified IgG may remain the only usable material. In these cases, researchers need primary structure evidence derived directly from the antibody protein rather than from amplifiable nucleic acids.
Antibody protein sequencing answers that need by using mass spectrometry to reconstruct heavy-chain and light-chain variable regions from purified antibody. The workflow digests IgG into peptides, acquires high-resolution LC-MS/MS spectra, interprets peptide fragmentation patterns, and assembles overlapping sequence evidence into VH and VL contigs with CDR annotation. Unlike database-assisted identification, which confirms a known reference, protein-level antibody sequencing can recover variable-region sequence information when the correct reference is missing, uncertain, or unavailable.
This article focuses on the technical logic of variable-region reconstruction: how LC-MS/MS generates peptide evidence, how de novo interpretation builds sequence tags, and how overlapping peptides are assembled into VH and VL sequences suitable for rescue, verification, and documentation.
Related Services
| Recommended Service | |
|---|---|
| Antibody protein sequencing | |
| MS-based antibody sequencing | |
| Full antibody sequencing | |
| IgG sequencing | |
| Heavy and light chain sequencing | |
| Protein-level confirmation |
Researchers evaluating antibody protein sequencing for VH/VL recovery can consult MtoZ Biolabs to review sample purity, isotype information, and expected variable-region coverage before LC-MS/MS analysis begins.
What Variable-Region Reconstruction Means in Practice
Antibody protein sequencing at the variable-region level asks a focused question: what VH and VL amino acid sequences are supported by peptide evidence obtained from this purified antibody?
The answer is built from many short peptide measurements rather than from one continuous readout. Immunoglobulin variable regions contain framework segments and complementarity-determining regions (CDRs) with higher sequence diversity than many other protein domains. CDR3 in particular can be short, modified, or difficult to cover with a single protease. Reliable reconstruction therefore depends on generating enough overlapping peptides across both chains to connect sequence tags into contiguous VH and VL regions.
The most important recovered outputs are usually:
When viable hybridoma cells or high-quality RNA remain available, hybridoma sequencing may be faster. When only purified antibody remains, mass spectrometry-based reconstruction becomes the primary route to variable-region sequence evidence.
Core Principles of MS-Based Variable-Region Reconstruction
1. From Purified IgG to Peptides
Antibody protein sequencing begins with purified IgG or another antibody preparation with acceptable purity and amount. The sample is typically reduced and alkylated to open disulfide-linked chains for digestion. One or more proteases then cleave the antibody into peptides suited to LC-MS/MS analysis.
Digestion design strongly affects variable-region coverage. Trypsin is widely used, but antibody variable regions may contain cleavage sites that leave CDR-adjacent regions underrepresented in a single digest. Complementary proteases can improve overlap across framework and CDR boundaries.
2. LC-MS/MS Acquisition and Peptide Fragmentation
After digestion, peptides are separated by liquid chromatography and analyzed by tandem mass spectrometry. Each selected precursor ion is fragmented to produce an MS/MS spectrum containing sequence-specific fragment ions. In peptide-centric sequencing, b-ions and y-ions are especially important because they support residue-by-residue interpretation of short sequence tags.
For antibody projects, acquisition goals include obtaining high-quality MS/MS spectra across both heavy-chain and light-chain peptides, with sufficient depth to support low-abundance or CDR-adjacent peptides when needed.
3. De Novo Peptide Interpretation
In variable-region reconstruction, many peptides cannot be assigned confidently by database search alone. De novo interpretation reads fragmentation patterns directly from the spectrum to derive sequence tags without requiring a trusted reference match.
Analysts prioritize spectra with clear fragmentation ladders, resolve ambiguities caused by isobaric residues when possible, and account for modifications that shift peptide mass or fragment patterns. For antibodies, homologous framework regions and somatic diversity increase the need for manual review.
4. Overlap Assembly into VH and VL Contigs
Individual peptide tags are not the final deliverable. Reconstruction proceeds by aligning overlapping peptides into longer contiguous regions on the heavy chain and light chain separately. A strong VH or VL assembly usually contains multiple independent overlapping peptides, consistent chain assignment, and explicit notation of unsupported intervals.
CDR annotation is then applied to the assembled variable-region sequences. Expert review remains important because one weak CDR peptide can disproportionately affect confidence in antigen-binding region assignment.
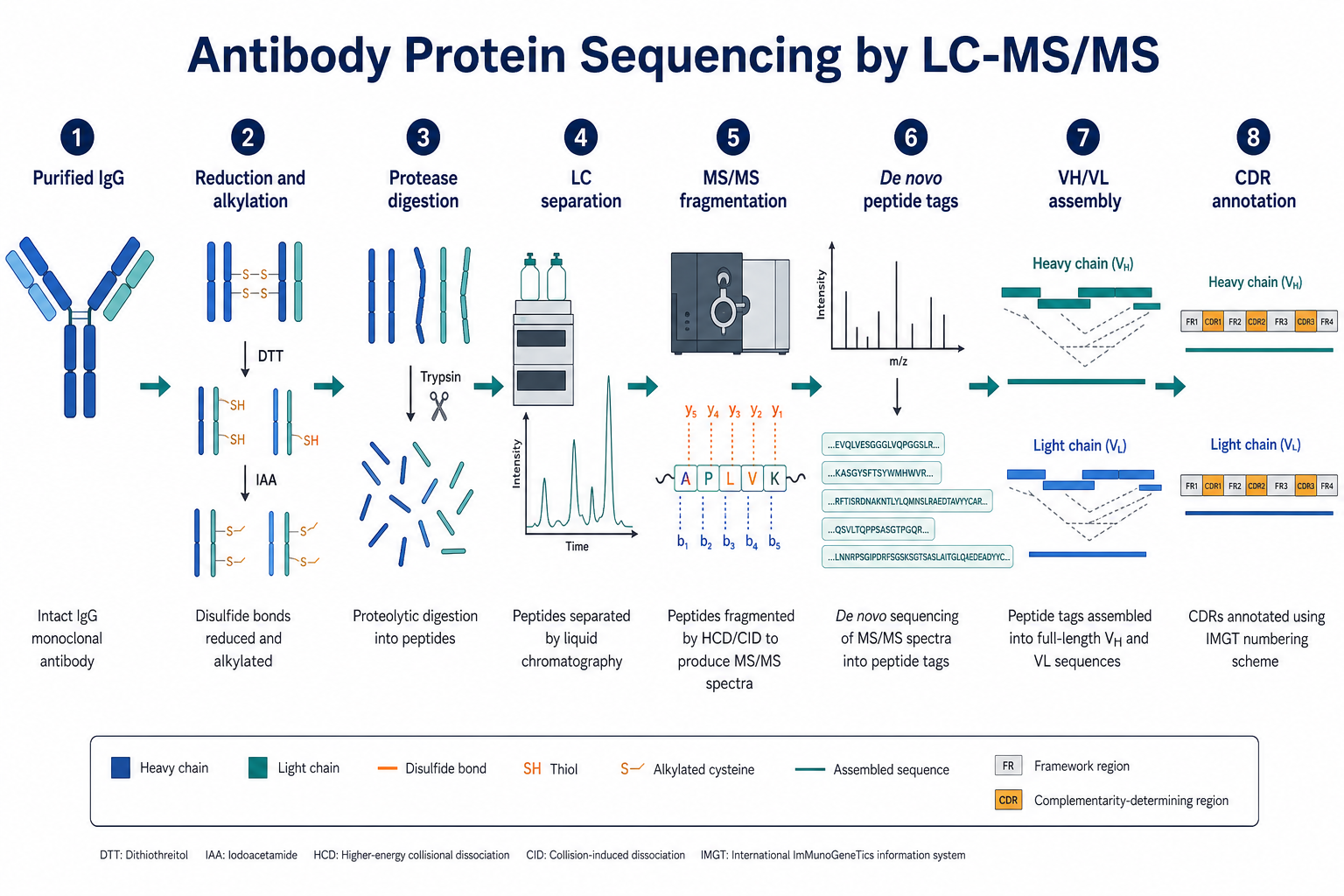
Figure 1. Antibody protein sequencing converts purified IgG into VH and VL sequence evidence through digestion, LC-MS/MS, peptide interpretation, and variable-region assembly.
Database-Assisted Matching vs De Novo Reconstruction for Antibodies
Database-assisted peptide mapping is efficient when the correct antibody sequence is already known and the goal is confirmation. Variable-region reconstruction for unknown, legacy, or undocumented antibodies usually requires de novo interpretation because the measured sequence may not be represented accurately in available databases.
| Approach | Best Use in Antibody Projects | Main Strength | Main Limitation |
|---|---|---|---|
| Database-assisted peptide mapping | Recombinant QC or comparability when reference is trusted | Fast confirmation against known VH/VL | Weak for unknown or divergent variable regions |
| De novo peptide sequencing | Legacy IgG rescue and undocumented antibody recovery | Works without a trusted reference | Requires strong spectra and expert assembly |
| Hybrid workflow | Partial reference with suspected CDR differences | Efficient use of both search and de novo paths | Needs clear rules for unmatched spectra |
| Multi-enzyme LC-MS/MS depth | VH/VL reconstruction with CDR coverage goals | Broader overlap across variable regions | Higher sample and analysis demand |
This distinction matters because antibody variable regions are often absent, incomplete, or incorrect in public databases even when constant-region information is available.
Digestion Strategies That Support VH/VL Coverage
Variable-region reconstruction is sensitive to protease choice. A single digestion strategy may leave CDR-adjacent or framework boundaries underrepresented, especially when peptides are too short, too long, or poorly ionized for high-quality MS/MS.
| Typical Role in Antibody Sequencing | Coverage Advantage | Common Risk | |
|---|---|---|---|
| Trypsin alone | Standard first-pass digest | Efficient for many IgG peptides | May miss useful overlap in some variable-region contexts |
| Chymotrypsin complement | Secondary digest for overlap | Can improve coverage across hydrophobic or CDR-adjacent regions | More complex interpretation |
| Glu-C or Lys-C complement | Targeted overlap expansion | Helps connect framework and CDR segments | Requires additional LC-MS/MS time |
| Multi-enzyme workflow | VH/VL reconstruction projects | Highest chance of contiguous assembly | Greater sample and analyst demand |
| Repeat LC-MS/MS acquisition | Rescue of weak variable-region coverage | Improves low-abundance peptide detection |
The best digestion plan depends on antibody isotype, sample amount, prior coverage results, and whether the project requires expression-ready CDR annotation or broader documentation only.
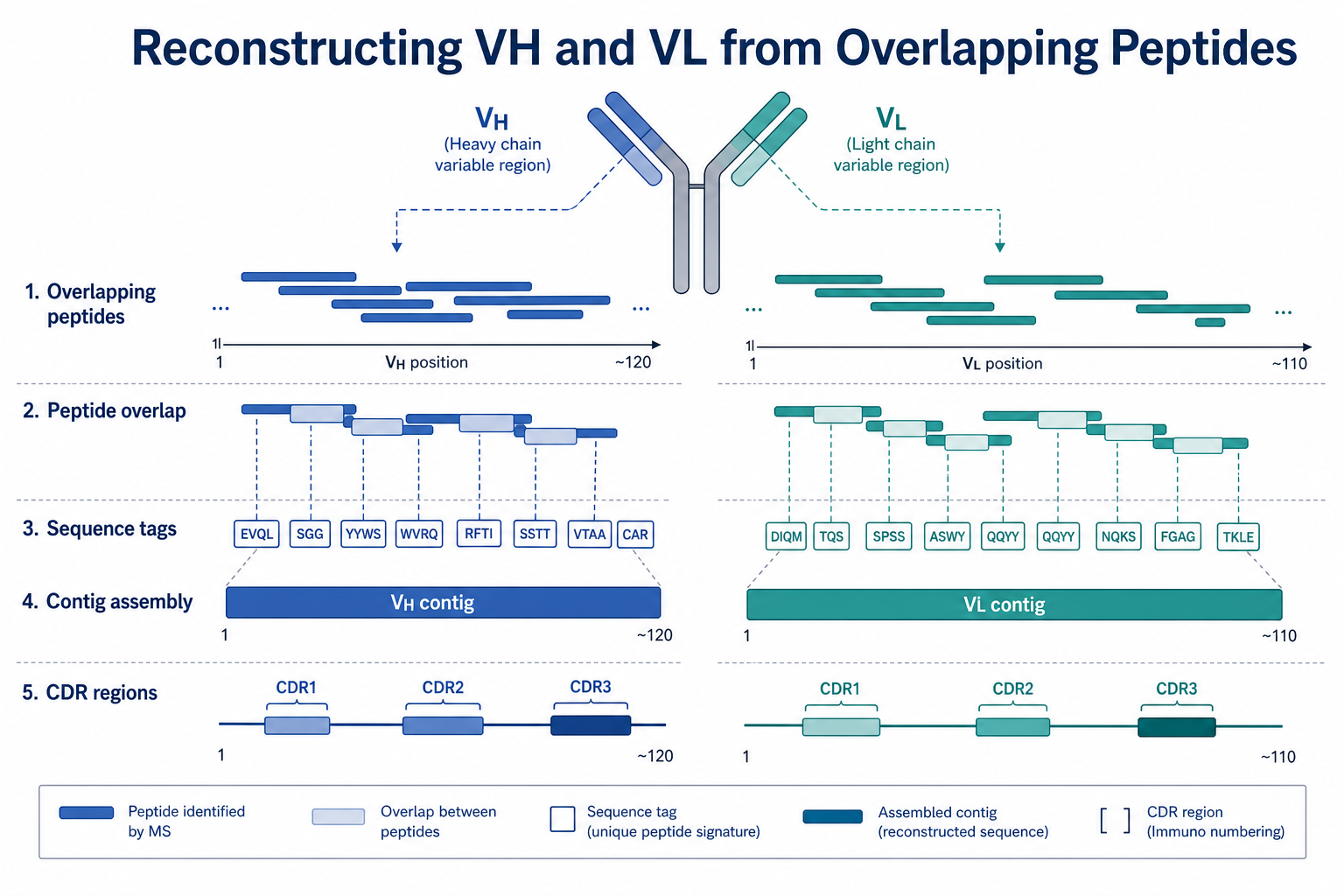
Figure 2. VH and VL reconstruction depends on overlapping peptide evidence assembled into contiguous variable-region sequences with CDR annotation.
Standard LC-MS/MS Workflow for Variable-Region Recovery
A robust antibody protein sequencing project usually follows a defined sequence of steps. Each step affects variable-region coverage, CDR confidence, and report usability.
1. Sample Feasibility Review
Assess antibody purity, amount, buffer compatibility, isotype, and expected reporting goal.
2. Sample Preparation
Reduce, alkylate, and digest IgG using one or more proteases matched to coverage needs.
3. LC-MS/MS Acquisition
Generate high-resolution fragmentation data across heavy-chain and light-chain peptides.
4. Spectrum Interpretation
Assign peptides by de novo analysis, database support, or a hybrid strategy as appropriate.
5. Chain-Specific Assembly
Build VH and VL contigs from overlapping peptide evidence on each chain.
6. CDR Annotation and QC Review
Assign CDR boundaries, flag ambiguous residues, and document unsupported regions.
7. Report Delivery
Provide annotated sequences, coverage maps, and validation recommendations.
Sample purity strongly affects outcome. A dominant IgG preparation can support high-confidence variable-region reconstruction. A complex mixture may require additional purification before meaningful VH/VL assembly is possible.
Core Advantages and Current Limitations
1. Core Advantages
(1) Works from purified antibody protein. Variable-region reconstruction does not require viable hybridoma cells, RNA, or expression plasmids.
(2) Direct protein-level evidence. The sequence is derived from the antibody molecule itself, which is valuable for legacy rescue and recombinant verification.
(3) Suitable for undocumented antibodies. De novo LC-MS/MS interpretation can recover VH/VL information when no trusted reference exists.
(4) Compatible with orthogonal confirmation. Peptide mapping, intact mass analysis, and CDR-focused follow-up can strengthen the final report.
2. Current Limitations
(1) Coverage is not automatic. CDR regions, especially CDR3, may require multi-enzyme design and repeat LC-MS/MS acquisition.
(2) Isobaric residues create ambiguity. Leucine and isoleucine may not be distinguished by routine mass workflows alone.
(3) Glycosylation and heterogeneity add complexity. Fc glycosylation and sample heterogeneity can affect peptide detectability and review time.
(4) Not always the fastest route when genetic material remains. Hybridoma sequencing may be more efficient when healthy cells or RNA are still accessible.
Antibody protein sequencing is powerful, but variable-region confidence depends on sample quality, digestion design, spectral depth, and expert review.
Applications in Antibody Discovery and Characterization
Mass spectrometry-based variable-region reconstruction supports multiple research and development workflows.
1. Hybridoma Rescue When Cells Are Lost
Purified IgG may be the only remaining source of sequence information.
2. Recombinant Antibody Verification
Expressed products can be checked against intended VH/VL design or documented divergence.
3. Legacy Sample Recovery
Antibody material with incomplete records can still yield variable-region evidence when purity and amount are sufficient.
4. Primary Structure Documentation
Annotated VH/VL sequences support internal records, publication, and tech transfer.
5. Comparator and Biosimilar Support
Protein-level sequence evidence can complement functional and analytical comparability studies.
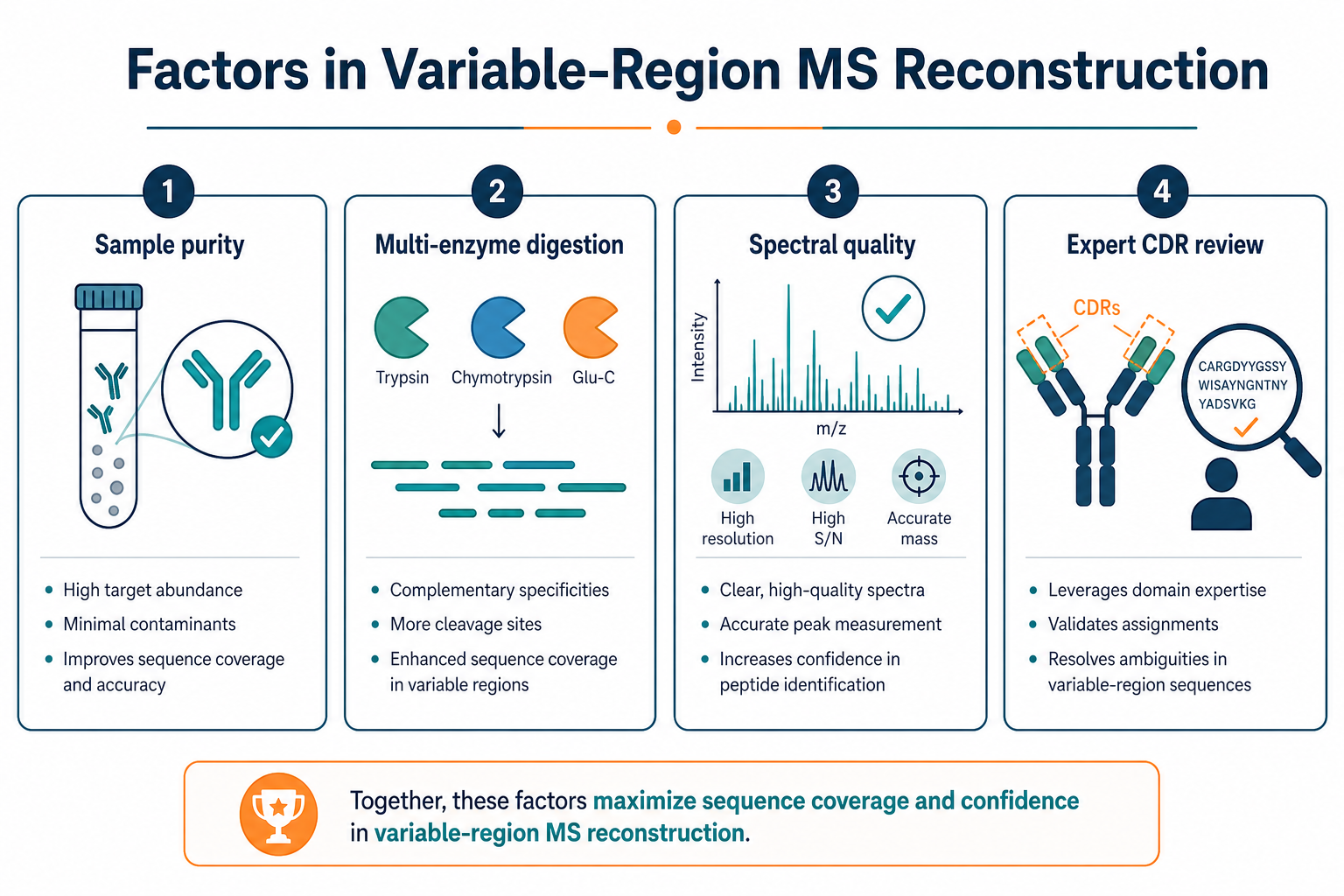
Figure 3. Variable-region reconstruction quality depends on sample purity, digestion design, spectral quality, and expert CDR review.
Factors That Affect Variable-Region Reconstruction Confidence
The infographic above summarizes the main practical drivers. The table below adds project-level detail for teams planning VH/VL recovery from purified antibody.
| What It Influences | Practical Implication | |
|---|---|---|
| Antibody purity | Peptide specificity and assembly clarity | Cleaner IgG improves VH/VL confidence |
| Sample amount | Depth of LC-MS/MS and repeat analysis | Limited material may restrict multi-enzyme design |
| Isotype and chain pairing | Digestion pattern and interpretation complexity | Metadata should be documented before analysis |
| Protease selection | Overlap across framework and CDR regions | Multi-enzyme workflows often improve reconstruction |
| Spectral quality | Strength of de novo sequence tags | Weak MS/MS may require repeat acquisition |
| CDR complexity | Confidence in antigen-binding region assignment | CDR3 may need targeted follow-up |
| Modifications | Mass shifts and fragment pattern changes | Glycosylation and other PTMs should be considered in review |
| Expert manual review | Final confidence in VH/VL and CDR calls |
These factors explain why two purified antibody samples with similar apparent purity can produce very different variable-region reconstruction outcomes.
Validation and Quality Considerations
A variable-region sequence report should be evaluated by peptide coverage, replicate support, chain assignment logic, and biological plausibility. Useful validation steps may include:
Transparent reporting improves usability for vector design and downstream discovery decisions. Low-confidence CDR segments should be treated cautiously when the sequence will be used directly for expression construct design.
Future Outlook
Antibody protein sequencing continues to benefit from improved MS sensitivity, better de novo interpretation tools, and more efficient multi-enzyme workflows for variable-region coverage. Laboratories are increasingly using protein-level reconstruction not only for rescue, but also as an orthogonal check on recombinant products and legacy materials. At the same time, expert review remains important because antibody variable regions are diverse, somatically mutated, and sensitive to sample-specific constraints.
For many teams, outsourcing antibody protein sequencing provides access to optimized digestion strategies, high-resolution LC-MS/MS, and reporting formats suited to rescue, verification, or documentation needs without building every capability internally.
Frequently Asked Questions
1. How does mass spectrometry reconstruct VH and VL sequences?
LC-MS/MS measures fragmented peptides from digested IgG. De novo interpretation derives sequence tags from MS/MS spectra, and overlapping peptides are assembled into VH and VL contigs with CDR annotation.
2. Is antibody protein sequencing the same as peptide mapping?
Not exactly. Peptide mapping confirms agreement with a known reference. Variable-region reconstruction can recover sequence information when the reference is missing or unreliable.
3. Can this method work when hybridoma cells are lost?
Yes, if purified antibody with acceptable purity and amount remains available. This is one of the main reasons teams use protein-level antibody sequencing.
4. Does antibody protein sequencing always recover full CDR coverage?
Not automatically. CDR coverage depends on digestion design, spectral quality, and sample amount. CDR3 in particular may require additional analysis.
5. Can LC-MS/MS distinguish leucine and isoleucine in antibody sequences?
Routine workflows often cannot distinguish these isobaric residues by mass alone. Reports should note this limitation when relevant.
Conclusion
Antibody protein sequencing reconstructs VH and VL variable-region sequences from purified antibody by combining protease digestion, LC-MS/MS acquisition, de novo peptide interpretation, overlap assembly, and CDR annotation. It provides direct protein-level evidence when genetic source material is unavailable and supports rescue, verification, and documentation workflows that depend on variable-region primary structure. The strongest outcomes come from matching digestion and acquisition depth to coverage needs, reviewing CDR assignments carefully, and validating the final sequence for the intended downstream use. Researchers planning antibody protein sequencing for VH/VL recovery can contact MtoZ Biolabs to review sample readiness, define coverage goals, and align the LC-MS/MS workflow with rescue, expression, or documentation requirements.
How to order?

