





Workflow for TMT 16plex Quantitative Proteomics Analysis
-
High throughput: up to 16 samples can be simultaneously analyzed in a single experiment.
-
High reproducibility: technical variation is significantly minimized.
-
Broad applicability: compatible with complex biological matrices such as tissues, cultured cells, serum, and cerebrospinal fluid.
-
Strong data integration capability: particularly suitable for clinical cohorts and multi–time-point experimental designs.
-
RIPA buffer or SDS buffer combined with ultrasonic lysis is commonly recommended.
-
Protein concentration is typically determined using the BCA assay to ensure equal protein input across samples.
-
Trypsin digestion is commonly performed at an enzyme-to-protein ratio of 1:50 and incubated overnight at 37°C.
-
Resulting peptides are desalted using C18 columns to remove interfering substances.
-
A labeling efficiency exceeding 95% should be ensured for all samples.
-
Prior to pooling, samples must be combined at equal ratios to prevent quantitative bias.
-
Depending on experimental objectives, 8-24 fractions may be generated.
-
Fractionation effectively reduces sample complexity and improves the detection of low-abundance proteins.
-
LC gradients are typically set to 90-120 minutes to balance chromatographic separation and analytical throughput.
-
MS3-based acquisition strategies (such as SPS-MS3) effectively mitigate ratio compression of TMT reporter ions and enhance quantitative accuracy.
-
Protein identification confidence is commonly controlled at a false discovery rate (FDR) of 1%.
-
Functional annotation and pathway enrichment analyses are performed using GO, KEGG, and protein-protein interaction (PPI) network construction.
-
Advanced analyses, including differential expression screening, clustering analysis, and machine learning-based modeling, can be conducted.
-
Multiple high-end mass spectrometry platforms: including Orbitrap Exploris 480, Fusion Lumos, and QE HF.
-
In-house optimized fractionation and enrichment strategies: enabling improved detection of low-abundance proteins.
-
An experienced data analysis team: supporting in-depth data mining and visualization-ready reporting.
-
Flexible collaboration models: ranging from pilot single-sample studies to large-scale projects involving hundreds of samples.
In systems biology research, accurate protein quantification is essential for characterizing the dynamic regulation of biological processes. Although traditional label-free approaches are relatively straightforward to implement, they often suffer from limitations in data consistency and control of batch effects. Owing to its high throughput, reduced inter-batch variability, and superior quantitative accuracy, Tandem Mass Tag (TMT) labeling has emerged as one of the most widely adopted strategies for multi-sample quantitative proteomics.
Overview of the Technical Principles of TMT 16plex
TMT (Tandem Mass Tag) is a chemical labeling strategy based on isobaric tags. All TMT tags exhibit identical masses at the MS1 level; however, upon MS2 fragmentation, they generate reporter ions with distinct masses, thereby enabling multiplexed quantitative analysis.
Advantages of TMT 16plex include:
Detailed Description of the Complete Experimental Workflow
1. Sample Protein Extraction and Quantification
High-quality protein extraction constitutes the foundation of reliable quantitative proteomics. Appropriate lysis buffers should be selected according to sample type to ensure high protein recovery, minimal degradation, and low levels of contaminants.
2. Protein Reduction, Alkylation, and Enzymatic Digestion
Proteins are subjected to reduction with dithiothreitol (DTT) and alkylation with iodoacetamide (IAA) to disrupt disulfide bonds and block free thiol groups, thereby facilitating efficient tryptic digestion.
3. TMT 16plex Labeling Reaction
Each sample is individually dissolved in TMT reagent solvent and reacted with the corresponding tryptic peptides. Labeling is completed within 30–60 minutes, and labeling efficiency can be evaluated using LC-MS or test strips.
4. High-pH Reversed-Phase Fractionation (High-pH RPLC)
Because ion suppression effects in mass spectrometry limit the number of peptides detectable per injection, high-pH reversed-phase fractionation is typically applied after TMT sample pooling to enhance proteome coverage and quantitative depth.
5. LC-MS/MS Analysis (Mass Spectrometry Detection)
The selection of the mass spectrometry platform critically determines analytical sensitivity and overall data quality. For TMT 16plex experiments, Orbitrap-based instruments with high resolution and fast scan rates (e.g., Exploris 480 and Fusion Lumos) are generally recommended.
6. Data Analysis and Bioinformatics Mining
Raw mass spectrometry data are processed for protein identification and quantification using software platforms such as Proteome Discoverer and MaxQuant, followed by integrative analysis in conjunction with multi-omics datasets or clinical metadata.
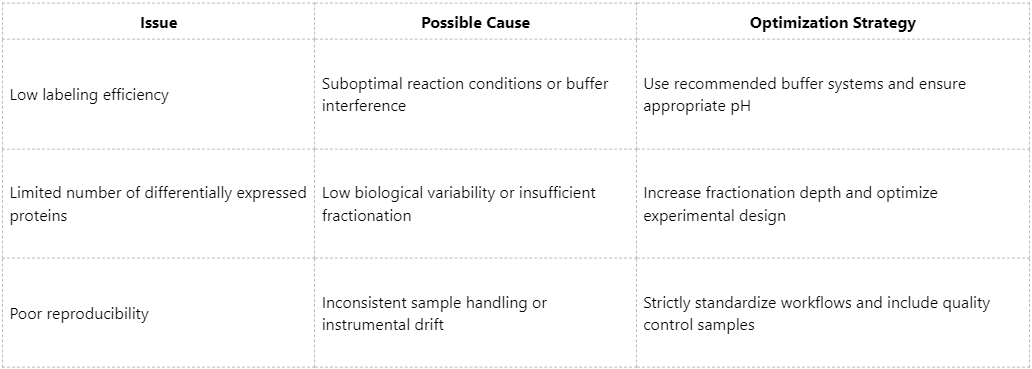
Common Issues and Optimization Strategies
Advantages of TMT Quantitative Proteomics Services at MtoZ Biolabs
As a professional provider of life science research services, MtoZ Biolabs has established an integrated platform encompassing TMT experimental design, sample preparation, mass spectrometry analysis, data processing, and biological interpretation.
Key advantages include:
With its high throughput and quantitative precision, TMT 16plex technology is increasingly regarded as a standard approach in quantitative proteomics. Careful optimization at every stage-from sample preparation to data interpretation-is essential for ensuring robust and reproducible results. MtoZ Biolabs is committed to supporting the scientific community by delivering high-quality proteomics data with strong biological interpretability and reproducibility, and welcomes collaborative efforts to advance precision life science research.
MtoZ Biolabs, an integrated chromatography and mass spectrometry (MS) services provider.
Related Services
How to order?

