





What Is the Workflow of Histone Phospho-Proteomics?
-
Common sources include cell lines, tissue samples, and blood cells.
-
Samples should be rapidly processed under low-temperature conditions to minimize interference from kinase and phosphatase activities and preserve the native modification states.
-
Acid extraction: Histones can be efficiently extracted using acid-based methods, typically with 0.4 N H₂SO₄ or HCl for nuclear protein treatment.
-
Commercial kits: These approaches improve reproducibility and operational efficiency.
-
A combination of trypsin and Glu-C is commonly employed to enhance peptide coverage.
-
Chemical derivatization (e.g., propionylation) is typically performed prior to digestion to neutralize the strong basicity of histones.
-
Nano-flow LC with low-pH gradients is recommended to enhance separation performance.
-
Data acquisition strategies include DDA (data-dependent acquisition) and DIA (data-independent acquisition).
-
Technical replicates and isobaric labeling-based quantification (e.g., TMT or iTRAQ) are recommended to improve quantitative robustness.
-
Identification of phosphorylation sites and modification counts.
-
Statistical analysis of differential phosphorylation sites between groups.
-
Pathway enrichment analysis (e.g., GO, KEGG).
-
Functional prediction of key modification sites (e.g., involvement in chromatin remodeling and transcriptional activation).
Histone phosphorylation is a critical epigenetic modification that is extensively involved in chromatin remodeling, gene transcription, DNA damage response, and other biological processes. Leveraging high-resolution mass spectrometry, researchers can systematically characterize the dynamic changes of histone phosphorylation sites and elucidate their regulatory roles in cell cycle progression, tumorigenesis, and immune responses. As a key branch of histone modification omics, histone phosphorylation has emerged as an active area of research in epigenetics.
Sample Processing and Histone Extraction
Histones are predominantly localized in the nucleus and mainly include H1, H2A, H2B, H3, and H4. Phosphorylation typically occurs on serine (Ser), threonine (Thr), or tyrosine (Tyr) residues located in their N-terminal tails.
1. Sample Type Selection
2. Extraction Strategy
Histone Digestion and Phosphopeptide Enrichment
Due to their low molecular weight and high basicity, histones are not well suited to conventional proteolytic workflows and require tailored sample preparation strategies.
1. Digestion Step
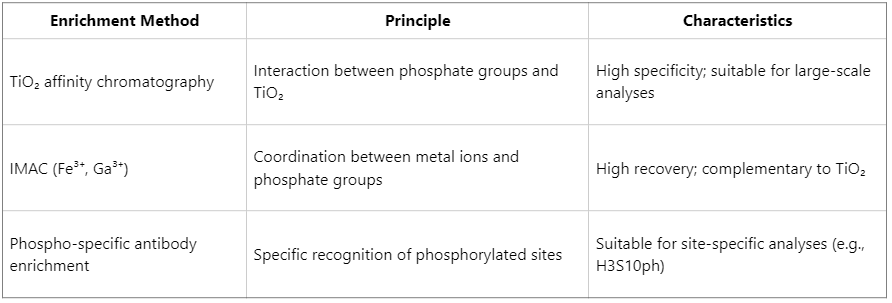
2. Phosphopeptide Enrichment Methods
The inherently low abundance of phosphorylation necessitates efficient enrichment strategies:
High-Resolution Mass Spectrometry Analysis
Following phosphopeptide enrichment, LC–MS/MS analysis is performed, typically using high-resolution Orbitrap or Q-TOF platforms.
Technical Considerations
Bioinformatics Analysis and Functional Interpretation
After processing raw mass spectrometry data using search engines (e.g., MaxQuant, Proteome Discoverer), the following information can be obtained:
Integration of histone variant information with site-specific analyses facilitates the construction of regulatory networks underlying chromatin state dynamics.
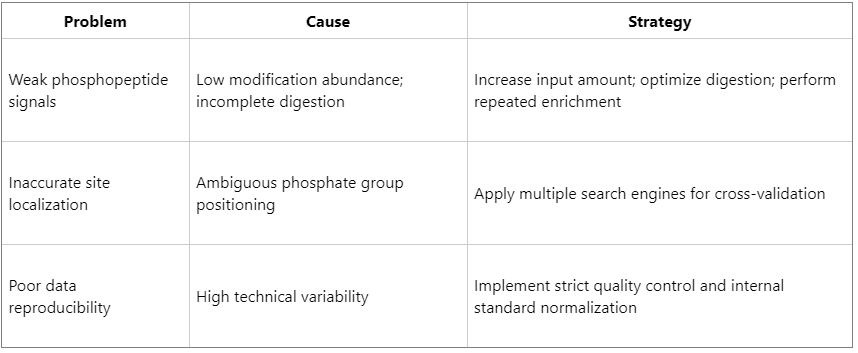
Common Challenges and Optimization Strategies
Histone phosphorylation represents a dynamic epigenetic mark. Systematic characterization of its modification landscape using proteomics approaches provides critical insights into the molecular mechanisms governing chromatin state transitions, cell fate determination, and tumorigenesis. MtoZ Biolabs has extensive expertise in histone modification omics and offers comprehensive, one-stop solutions, including histone extraction, phosphopeptide enrichment, quantitative mass spectrometry, and bioinformatics analysis, thereby enabling efficient exploration of epigenetic regulatory mechanisms.
MtoZ Biolabs, an integrated chromatography and mass spectrometry (MS) services provider.
Related Services
How to order?

