





What Are the Quantitative Methods for Proteomics?
- SILAC (metabolic labeling)
- TMT, iTRAQ (isobaric chemical labeling)
- Dimethyl (chemical methylation labeling)
- AQUA, MRM, PRM (targeted labeling or internal standard-based quantification)
- No specialized reagents are required, resulting in relatively low experimental cost
- No inherent limitation on sample number, offering high scalability
- Well suited for complex sample types such as tissues and serum
- Highly dependent on mass spectrometry stability, requiring rigorous inter-batch normalization
- Lack of internal reference standards makes it difficult to fully eliminate systematic bias
- Preliminary differential screening of clinical samples
- Exploratory analysis of global protein expression patterns
- Can be combined with post-translational modification-focused analyses (e.g., phosphoproteomics)
- Stable isotope-labeled amino acids containing ¹³C and/or ¹⁵N (e.g., ¹³C₆-Arg, ¹³C₆-Lys) are supplemented into cell culture media.
- After several generations of cell proliferation, newly synthesized proteins become fully metabolically labeled.
- Labeled and unlabeled cell populations are mixed prior to enzymatic digestion and mass spectrometry analysis, thereby minimizing experimental variability.
- High labeling efficiency enables accurate and reliable quantification
- Early-stage sample mixing reduces technical variation
- Well suited for dynamic studies such as protein synthesis and degradation
- Restricted to cell culture systems and not applicable to animal or tissue samples
- Requires extended culture periods and incurs higher costs compared with LFQ
- Time-course studies of drug treatment effects
- Kinetic analysis of post-translational modification dynamics
- Analysis of tumor cell responses to external stimuli
- Isobaric tags are used to chemically label peptides following enzymatic digestion.
- All labeled peptides exhibit identical precursor masses at the MS1 level, while distinct reporter ions are released during MS2 fragmentation.
- Relative quantification across multiple samples is achieved by comparing the intensities of reporter ions.
- Supports multiplexed analysis of multiple samples in parallel (typically 6-18 samples per batch)
- Low inter-batch variability, making it suitable for large-scale experimental designs
- High compatibility with high-resolution mass spectrometry platforms such as Orbitrap
- Quantitative accuracy may be affected by ratio compression effects
- Relatively high experimental cost due to the expense of labeling reagents
- Differential protein expression analysis between disease and control groups
- Experimental designs involving multiple conditions or time points
- Quantitative analysis of samples from multiple tissues or biological replicates
- Formaldehyde (or deuterated formaldehyde) in combination with sodium cyanoborohydride (NaBH₃CN) is used to methylate peptide N-termini and lysine residues.
- This strategy generates light, medium, and heavy isotopically distinct peptides, enabling quantitative comparison of up to three sample groups.
- Low cost and rapid chemical reaction
- Labeling efficiency typically exceeds 95%
- Suitable for small- to medium-scale comparative studies
- Limited throughput due to a restricted number of labeling channels (generally ≤3)
- Potential quantitative bias may be introduced during sample mixing.
- Small-scale differential protein expression analysis
- Rapid exploratory or pilot experiments
- Quantitative analysis of modified peptides
- Performed on triple quadrupole mass spectrometers
- Specific precursor-fragment ion transitions are predefined
- Suitable for highly sensitive quantification of a limited number of target proteins
- Implemented on Orbitrap-class high-resolution mass spectrometers
- A single precursor ion triggers full-fragment ion acquisition
- Maintains the specificity of MRM while providing higher resolution and improved quantitative stability
- Synthetic stable isotope-labeled standard peptides are co-analyzed with endogenous peptides.
- Enables direct absolute quantification of target proteins, typically reported in fmol/ng.
- Extremely high sensitivity, particularly suitable for quantification of low-abundance proteins in clinical samples
- High data reproducibility
- Commonly applied during biomarker validation stages
- Not suitable for high-throughput screening, and requires careful design of target peptides.
- Demands precise matching of isotopic standards and dedicated method development.
- Preclinical and clinical biomarker validation studies
- Quantitative immunological assessment of peptide vaccines
- Target protein monitoring in pharmacokinetic (PK) studies
Quantitative proteomics is a central analytical approach for investigating changes in protein expression and the underlying regulatory mechanisms. In recent years, this field has rapidly evolved to encompass a broad range of high-resolution and high-sensitivity strategies. A clear understanding of the available protein quantification methods, their suitable experimental contexts, and appropriate selection criteria is therefore essential. In the following sections, we systematically summarize the major quantitative proteomics technologies currently in widespread use, with the aim of assisting researchers in establishing rational experimental strategies at early stages of study design, thereby ensuring data reproducibility, result reliability, and clearly defined differential outcomes.
Classification of Methods for Quantitative proteomics
Protein quantification approaches in proteomics can generally be divided into two major categories:
1. Label-Free Quantification (LFQ)
(1) No exogenous labels are introduced during sample preparation.
(2) Protein abundance is directly inferred based on peptide ion intensities or spectral counts.
2. Label-Based Quantification
(1) Stable isotope labels are introduced through metabolic or chemical labeling strategies.
(2) Representative methods include:
Detailed Overview of Mainstream Protein Quantification Methods: Principles, Features, and Application Scenarios
1. Label-Free Quantification (LFQ)
(1) Principle
Samples are independently subjected to enzymatic digestion and mass spectrometry analysis. Relative protein expression levels are inferred by comparing the ion intensities (peak areas) or spectral counts of identical peptides across different samples.
(2) Features
(3) Application Scenarios
2. SILAC (Stable Isotope Labeling by Amino Acids in Cell Culture)
(1) Principle
(2) Features
(3) Application Scenarios
3. TMT (Tandem Mass Tag) and iTRAQ (Isobaric Tag for Relative and Absolute Quantitation)
(1) Principle
(2) Features
(3) Application Scenarios
MtoZ Biolabs supports TMTpro 16plex technology, allowing up to 16 sample groups to be compared within a single experiment, thereby substantially improving quantitative efficiency and analytical throughput.
4. Dimethyl Labeling (Chemical Methylation Labeling)
(1) Principle
(2) Features
(3) Application Scenarios
5. Targeted Quantification Techniques: MRM/SRM, PRM, and AQUA
(1) Principle
MRM/SRM (Multiple Reaction Monitoring)
PRM (Parallel Reaction Monitoring)
AQUA (Absolute Quantification)
(2) Features
(3) Application Scenarios
Comparative Summary of Different Methods for Quantitative Proteomics
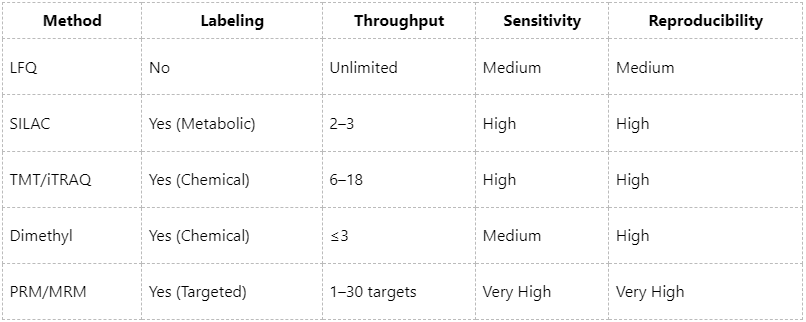
Recommended strategy: Perform global quantitative screening using TMT or LFQ, followed by PRM-based validation of key proteins, thereby establishing a closed-loop research workflow from discovery to verification.
How to Select an Appropriate Method for Quantitative Proteomics
The selection of an appropriate quantitative strategy depends not only on experimental budget but also on research objectives, sample types, available technical resources, and downstream validation requirements:

MtoZ Biolabs specializes in proteomics and mass spectrometry technologies and provides comprehensive, high-quality quantitative proteomics services covering the entire workflow from sample preparation to target validation. Supported by high-resolution mass spectrometry platforms such as Orbitrap Exploris 480 and Q Exactive Plus, MtoZ Biolabs offers a wide range of mainstream quantitative strategies, including TMT 6/10/16plex, iTRAQ 4/8plex, LFQ, SILAC, and PRM, all conducted under strict quality control standards to ensure data robustness and reliability. Service offerings encompass protein extraction, enzymatic digestion and labeling, mass spectrometry analysis, differential protein identification, GO and KEGG functional enrichment analysis, as well as PRM-based target validation, thereby addressing multi-level research needs from basic studies to preclinical development. These services are widely applied in mechanistic studies of cancer, cardiovascular and cerebrovascular diseases, neurological disorders, as well as in vaccine and antibody development, drug mechanism investigations, and biomarker validation, enabling researchers to efficiently obtain biologically meaningful quantitative proteomics data.
“Different protein quantification methods function as magnifying lenses for scientific questions; selecting the appropriate approach is the key to unlocking biological insight.” In the data-driven era of life sciences, quantitative proteomics extends beyond simply determining protein abundance. More importantly, it enables the elucidation of the biological logic and clinical relevance underlying changes in protein expression through precise and robust analytical strategies. For researchers designing proteomics experiments or seeking services for differential protein analysis and quantitative validation, MtoZ Biolabs provides individualized technical consultation, quantitative strategy recommendations, and experimental design evaluation to support further scientific advancement.
MtoZ Biolabs, an integrated chromatography and mass spectrometry (MS) services provider.
Related Services
How to order?

