





SWATH-MS Data Analysis Workflow and Common Pitfalls
-
Normalization by total intensity or median values (e.g., MSstats, Perseus)
-
Imputation of missing values (e.g., k-nearest neighbors, maximum likelihood estimation)
-
Differential expression analysis of proteins (e.g., Limma, t-test)
-
Functional annotation and enrichment analysis (e.g., GO, KEGG)
Introduction: The Power of SWATH-MS Relies on Proper Data Analysis
SWATH-MS (Sequential Window Acquisition of All Theoretical Fragment Ion Spectra), a representative technique in data-independent acquisition (DIA), has significantly advanced large-scale, highly reproducible, and label-free quantitative proteomics. However, the intrinsic complexity of the data poses challenges, including intricate analysis workflows, strong parameter dependencies, and high variability in analytical outcomes.
This article systematically presents the standard workflow for SWATH-MS data analysis, highlights the critical aspects of each step, and summarizes common pitfalls along with their corresponding solutions to assist researchers in avoiding the so-called "black hole of data analysis".
Overview of the Standard Workflow of SWATH-MS Data Analysis
SWATH-MS data analysis typically comprises the following five key steps:
1. Conversion of Raw Data to mzML Format
SWATH data are commonly generated by platforms such as SCIEX (.wiff) and Thermo (.raw). The first step involves converting these files into a standardized format using tools like ProteoWizard (e.g., msConvert) to enable compatibility with downstream analysis software.
(1) Notes: Ensure the data are maintained in centroid mode to avoid erroneous profile-mode conversions.
(2) Common Pitfalls: Failure to select appropriate conversion parameters may result in data redundancy or information loss.
2. Spectral Library Search
Accurate quantification in SWATH-MS is highly dependent on the use of high-quality spectral libraries. Commonly used tools include:
(1) OpenSWATH with PyProphet (open-source)
(2) DIA-NN (high processing speed, compatible with FASTA-based predicted libraries)
(3) Spectronaut (commercial software with a user-friendly graphical interface)
Peptide identification and quantification are achieved by matching fragment ions from SWATH acquisitions with those in the spectral library. MtoZ Biolabs provides high-quality spectral libraries for multiple species, as well as custom DDA library construction services, with support for user-uploaded reference databases.
3. FDR Control and Scoring Models (Statistical Scoring)
SWATH-MS data comprises hundreds of thousands of fragment ion peaks, which are susceptible to interference from background noise. Common scoring algorithms (e.g., PyProphet) generate multiple scoring features for each peptide (e.g., XCorr, retention time deviation, peak shape), which are then classified using machine learning models—typically semi-supervised LDA. Peptides are retained based on a false discovery rate (FDR) threshold, commonly set at 1%.
(1) It is recommended to apply a global FDR (global context) to achieve more robust peptide identification.
(2) Common pitfall: neglecting the quality of scoring model training and insufficient FDR control, which can result in false-positive identifications.
4. Retention Time Alignment (RT Alignment)
Peptide retention times may vary across different samples, necessitating alignment through algorithms such as TRIC or those integrated within Spectronaut. This process standardizes the detection time for identical peptides, thereby improving the precision of cross-sample comparisons.
(1) Incorporating internal standards (e.g., iRT) is recommended to enhance alignment accuracy.
(2) Pitfall: lack of alignment or failed alignment can cause quantification loss and inconsistent data points.
5. Data Normalization and Differential Analysis (Quantification & Stats)
Once the quantitative matrix is obtained, the following steps are typically applied:
(1) MtoZ Biolabs offers an integrated service combining differential protein analysis, data visualization, and pathway annotation.
(2) Pitfall: failing to perform batch correction before merging datasets from different experiments can introduce spurious differential results.
Common Pitfalls and Recommendations in SWATH-MS Data Analysis
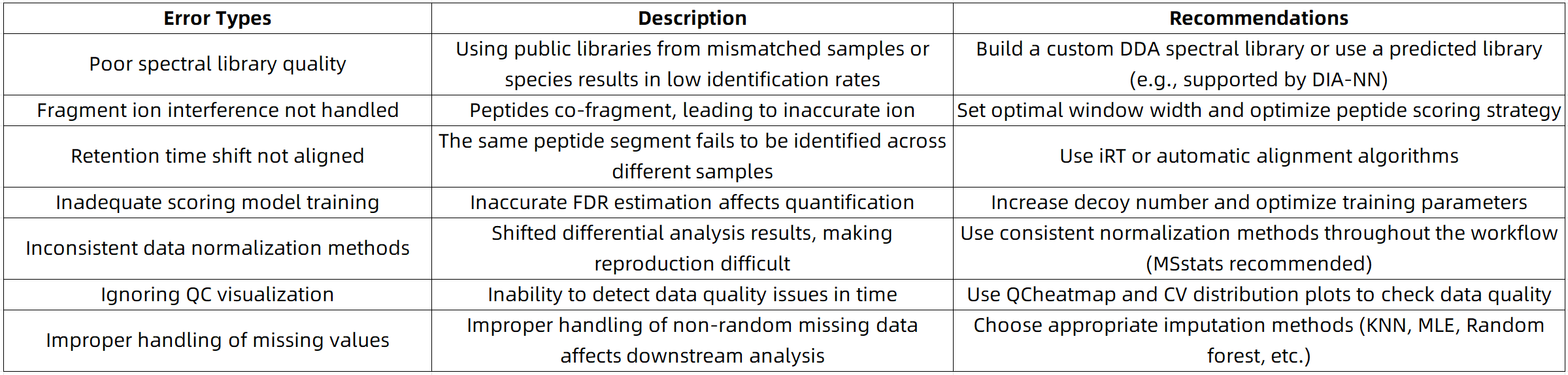
SWATH-MS Data Analysis Workflow Advantages from MtoZ Biolabs
To lower the technical barrier for users in SWATH-MS data analysis, we provide:
(1) Standardized SWATH-MS data processing workflows (OpenSWATH / DIA-NN / Spectronaut)
(2) Optimized spectral library generation, in silico spectrum prediction, and multi-species alignment strategies
(3) Normalization of the quantitative matrix, imputation of missing values, and identification of differentially expressed proteins
(4) Automated functional annotation, pathway enrichment analysis, and data visualization
Through platform-level optimization and support from our team of bioinformatics experts, we enable researchers to concentrate on their scientific investigations while entrusting the data analysis process to us with full confidence. The power of SWATH-MS lies not only in its unbiased data acquisition approach but also in the rigorous and specialized data analysis workflow that follows. Each step, though often considered a technical detail, plays a critical role in determining the depth of protein identification and the reliability of quantitative results. Only by thoroughly understanding and avoiding common pitfalls can the full scientific potential of SWATH-MS be realized. Please feel free to contact MtoZ Biolabs for a free evaluation of your SWATH-MS project and professional support in data analysis—we are committed to advancing every proteomic exploration with expertise.
MtoZ Biolabs, an integrated chromatography and mass spectrometry (MS) services provider.
Related Services
How to order?

