





SILAC-Based Quantitative PTM Analysis Service
SILAC-Based Quantitative PTM Analysis Service is a service that combines stable isotope labeling by amino acids in cell culture (SILAC) with high-resolution mass spectrometry to achieve relative quantification and precise site identification of PTMs under different treatment conditions. It is widely used in cutting-edge biomedical research areas such as signaling pathway studies, drug target screening, and mechanistic analysis of post-translational modifications.
Post-translational modifications (PTMs) are central regulatory mechanisms that control cellular signal transduction, metabolic homeostasis, and disease progression. These modifications include phosphorylation, acetylation, methylation, ubiquitination, and others. Dynamic quantitative analysis of these modifications is essential for understanding their biological functions and regulatory pathways.
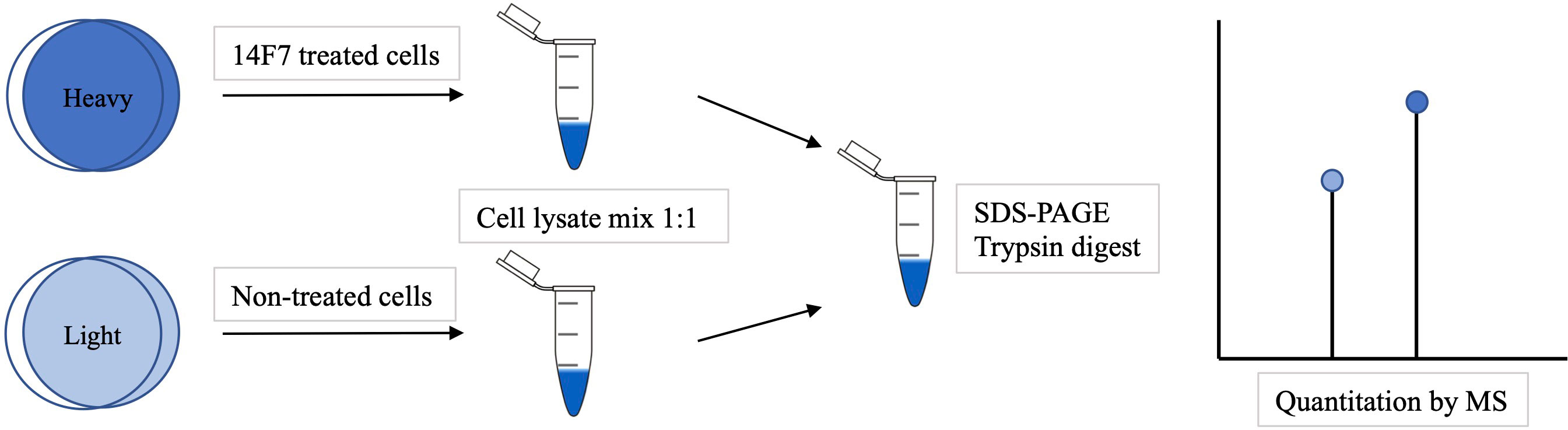
Bousquet PA. et al. Front Immunol. 2022.
SILAC is a quantitative proteomics technique based on metabolic labeling. It involves introducing light or heavy isotope-labeled lysine or arginine into the cell culture medium, allowing cells to incorporate these labeled amino acids during protein synthesis. Due to its ability to achieve high-precision and low-error relative quantification between samples, SILAC is widely used in quantitative PTM analysis. It is particularly suitable for condition-based comparisons, dynamic monitoring of modifications, and modification profiling in response to stimuli.
Leveraging advanced chromatography and mass spectrometry platforms, MtoZ Biolabs offers SILAC-Based Quantitative PTM Analysis Service enabling high-accuracy, low-bias quantification of post-translational modifications at the cellular level. This service helps researchers systematically analyze dynamic changes in PTMs, pinpoint critical functional sites, and uncover the biological significance of regulatory pathways under various treatment conditions or disease states.
Analysis Workflow
The main workflow of the SILAC-Based Quantitative PTM Analysis Service is as follows:
1. Cell Culture and SILAC Labeling
Cells from different treatment groups are cultured separately in light or heavy isotope-labeled amino acid media for at least six generations to ensure complete labeling.
2. Protein Extraction and Mixing
Cell lysates are collected and mixed in equal amounts, followed by unified digestion and subsequent processing.
3. Digestion and PTM Enrichment
Standard proteolytic digestion (e.g., trypsin) is performed, and enrichment strategies are selected based on the target PTM.
4. High-Resolution Mass Spectrometry Analysis
Peptides are analyzed using LC-MS/MS platforms to determine peptide mass and abundance.
5. Data Analysis
Quantitative software tools are used to calculate light/heavy peptide ratios, localize modification sites, and perform statistical analysis and functional enrichment.
Applications
SILAC-Based Quantitative PTM Analysis Service is suitable for a wide range of cellular models and research applications, including but not limited to:
Dynamic Signaling Pathway Studies
Precisely quantify phosphorylation or acetylation responses to reveal the functional networks of key kinases, acetyltransferases, or demodifying enzymes.
Drug Target Identification and Mechanism of Action Analysis
Compare PTM profiles before and after drug treatment to identify functional target proteins and regulatory patterns.
Mechanistic Studies in Disease Models
Analyze PTM changes in models of cancer, neurodegenerative diseases, and others to uncover disease-associated regulatory pathways.
Epigenetic and Chromatin Regulation Studies
Quantitatively assess histone acetylation, methylation, and other modifications involved in chromatin remodeling.
FAQ
Q. Does the Service Support Enrichment of Multiple PTM Types? How is Enrichment Specificity and Efficiency Ensured?
Yes. We support enrichment of various common PTMs, including phosphorylation, acetylation, methylation, and ubiquitination. Enrichment strategies are selected according to the PTM type—for example, TiO₂ and Fe³⁺-IMAC for phosphopeptides, and antibody affinity for acetylated or ubiquitinated peptides. Each enrichment workflow includes blank and positive controls, and we provide reports on peptide yield and PTM coverage before and after enrichment to ensure high specificity and low background interference.
Q. Can Relative Quantification be Achieved at the Modification Site Level rather than Just the Protein Level?
Absolutely. SILAC combined with high-resolution mass spectrometry enables quantification of individual peptides and even specific PTM sites, such as changes in phosphorylation at S473 or acetylation at K27. By applying customized search parameters and high-quality enrichment workflows, we can accurately detect and quantify dynamic changes at specific modification sites, providing direct evidence for PTM regulatory mechanisms.
Deliverables
1. Comprehensive Experimental Details
2. Materials, Instruments, and Methods
3. Total Ion Chromatogram & Quality Control Assessment (project-dependent)
4. Data Analysis, Preprocessing, and Estimation (project-dependent)
5. Bioinformatics Analysis
6. Raw Data Files
Case Study
This study employed a reversed-pulsed SILAC (rp-SILAC) strategy to systematically analyze the degradome, acetylome, and ubiquitinome under aspirin treatment. The results revealed that aspirin inhibited proteasome activity by inducing lysine acetylation of proteasome subunits, thereby reducing the degradation of soluble proteins. In addition, aspirin significantly increased K63-linked ubiquitination, which is associated with lysosomal degradation—particularly on α-synuclein (α-syn), a key protein in Parkinson’s disease. This K63-linked ubiquitination facilitated the clearance of α-syn aggregates and alleviated motor dysfunction in both cellular and mouse models of Parkinson’s disease.
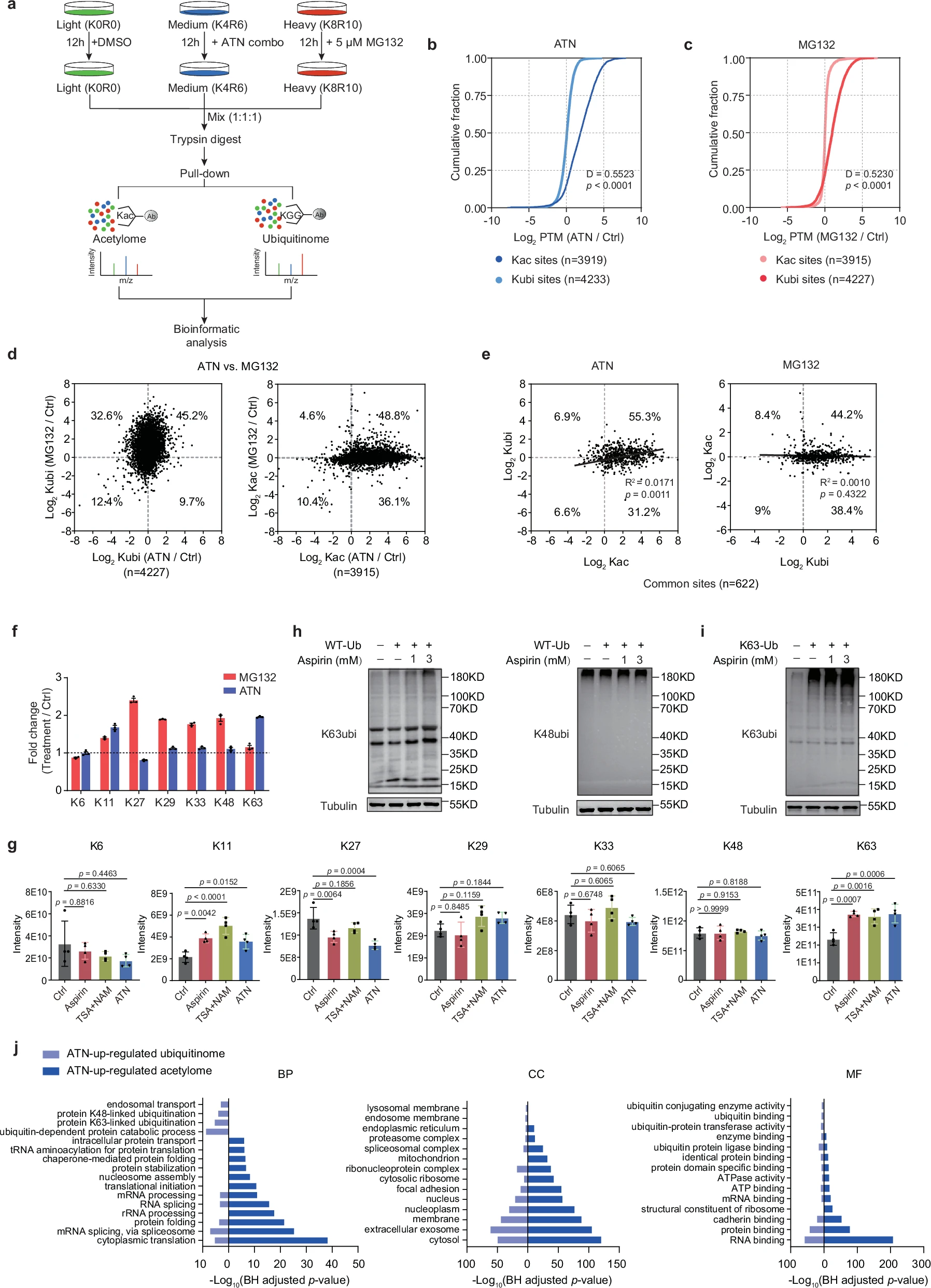
Gao J. et al. Nat Commun. 2025.
How to order?

