





Protein Sequencing Methods Compared: LC-MS/MS, Edman, N/C Terminal, and De Novo
Introduction
Choosing a protein sequencing method is rarely a simple technology preference. A researcher trying to identify an unknown gel band, a biologics team confirming primary structure, and a project manager recovering sequence information from a non-model organism all need sequence evidence. They do not need the same workflow.
The wrong method can create avoidable cost. Edman sequencing may be selected for a protein with a blocked N-terminus. Database-assisted LC-MS/MS may be used when the reference sequence is missing. Peptide mapping may be ordered when the true need is de novo sequence recovery. Each method is useful, but only when the sample and decision match the evidence it can produce.
The best protein sequencing strategy starts with four questions: what sample is available, how pure the sample is, what sequence information is already known, and what decision the result must support. If these answers are unclear, MtoZ Biolabs can compare method fit before researchers choose a route for limited or expensive protein samples.
Related Services
| Customer Need | Recommended Service Direction |
| Need broad method selection for sequence evidence | Protein Sequencing Service |
| Need MS-based sequence analysis | Protein Sequencing Service by Mass Spectrometry |
| Need N- or C-terminal sequence analysis | N/C Terminal Sequencing Service |
| Need sequence confirmation against a reference | Peptide Mapping Service |
| Need unknown or database-limited sequence recovery | De Novo Sequencing Service |
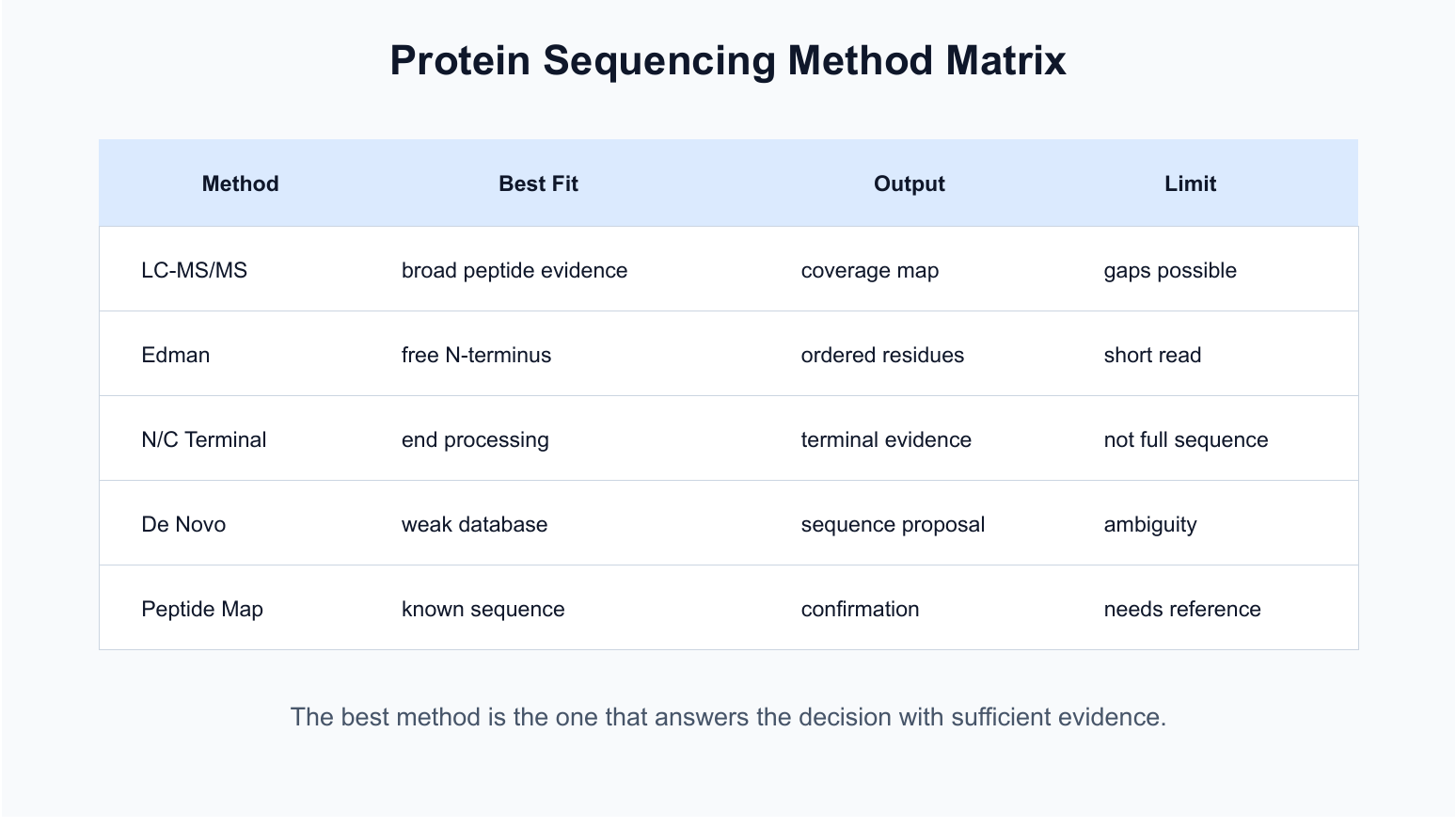
Figure 1. Protein sequencing methods differ by sample fit, evidence type, and limitation.
LC-MS/MS Protein Sequencing
LC-MS/MS protein sequencing is often the most flexible route for peptide-level sequence evidence. The protein is digested into peptides, separated by liquid chromatography, fragmented by tandem mass spectrometry, and interpreted through database search, peptide mapping, or de novo analysis.
The strength of LC-MS/MS is breadth. It can identify proteins, confirm sequence coverage, detect expected peptides, support variant investigation, and provide internal sequence information even when direct terminal reads are not available. It is especially useful for proteins that are too large or complex for a single direct read.
The limitation is that sequence coverage is not automatically complete. Some regions may digest poorly, ionize weakly, or fragment insufficiently. Hydrophobic segments, heavily modified regions, disulfide-linked peptides, and low-abundance fragments may be underrepresented. For high-confidence primary-structure work, method design and expert review matter as much as the instrument run.
Use LC-MS/MS protein sequencing when the project needs broad peptide evidence, identity confirmation, sequence coverage, or support for known and partially known proteins. It is often a good starting point when the sample can be digested and the result must be reusable in a report.
Edman Sequencing
Edman degradation reads amino acids sequentially from an accessible N-terminus. It is a classic method with a clear output when the chemistry works well. For purified proteins or peptides with a free N-terminus, it can provide direct residue information across a limited read length.
The main advantage is interpretability. A successful Edman read gives ordered N-terminal residues without relying on database matching. This can be valuable for confirming processing, cleavage, or the identity of a purified peptide.
The limitation is scope. Edman sequencing usually does not recover full-length protein sequence. It also struggles with blocked N-termini, mixtures, low sample amount, internal sequence needs, and proteins whose N-terminus is modified or inaccessible. If the protein has an N-terminal acetylation or pyroglutamate formation, direct Edman analysis may produce little usable sequence.
Use Edman sequencing when the specific question is an accessible N-terminal read from a sufficiently pure protein or peptide. Do not use it as the default choice for full protein sequencing.
N-Terminal and C-Terminal Sequencing
N-terminal and C-terminal sequencing focus on protein ends. Terminal information is important because processing events often occur at the ends of proteins. Signal peptide cleavage, propeptide maturation, tag removal, proteolytic truncation, degradation, and terminal heterogeneity can all affect biological interpretation or product quality.
N-terminal sequencing can be performed by Edman chemistry or MS-based approaches, depending on accessibility and project design. C-terminal sequencing is often more challenging and may require targeted MS strategies, enzymatic design, or comparison against expected terminal peptides.
Terminal sequencing is not the same as full sequence recovery. It is most useful when the end of the protein is the decision point. For example, a biologics team may need to confirm whether the expected C-terminal lysine is present or removed. A recombinant protein team may need to confirm whether a secretion signal was correctly processed.
Use terminal sequencing when end processing matters. If the project also needs internal coverage, combine terminal analysis with LC-MS/MS peptide mapping or broader protein sequence analysis.
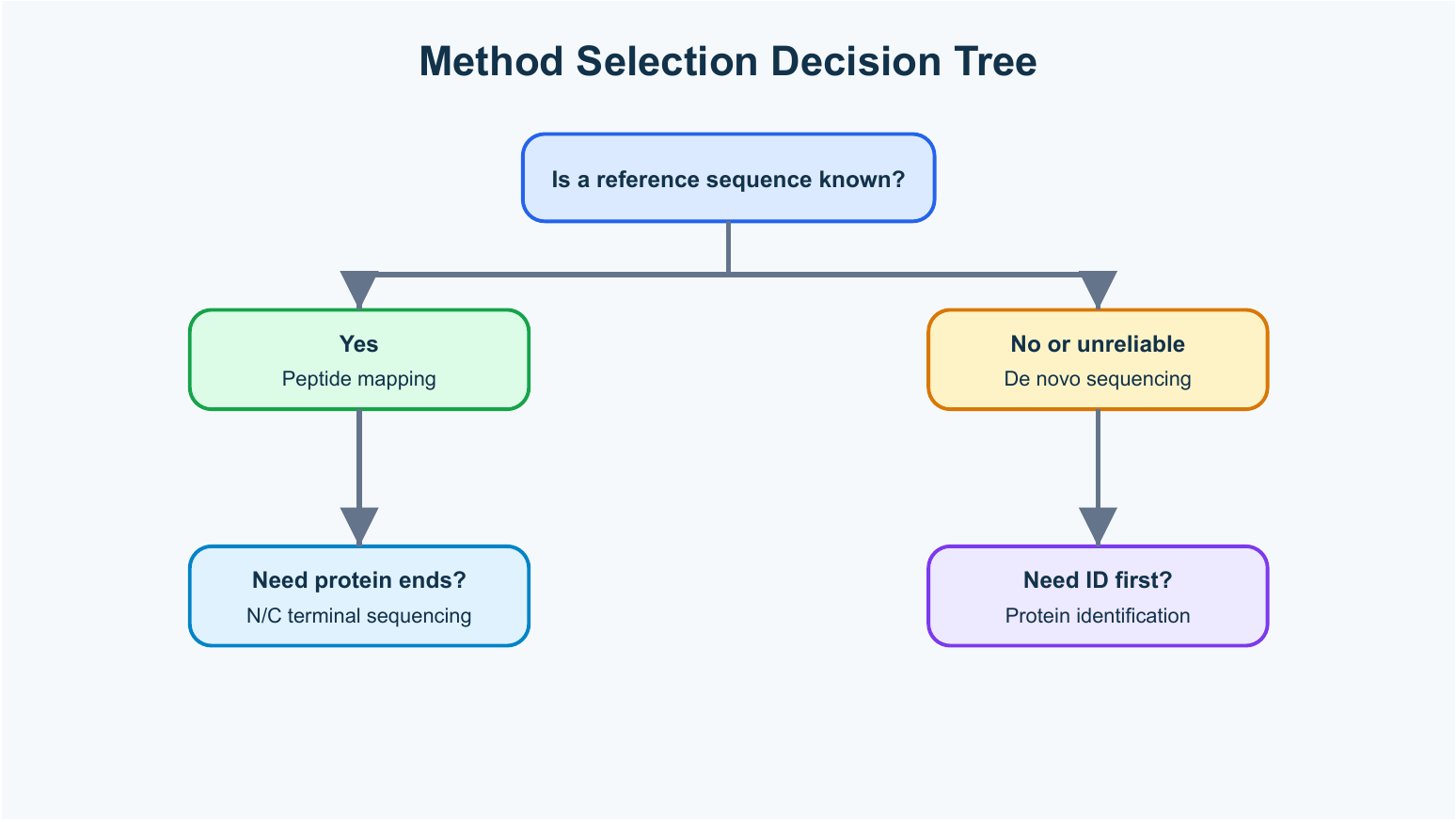
Figure 2. Method selection should follow sample type, known sequence information, and required evidence.
De Novo Protein Sequencing
De novo protein sequencing is used when sequence information must be inferred without depending fully on a database. In LC-MS/MS-based workflows, peptide fragmentation spectra are interpreted to propose amino acid sequences. Multiple enzymes, overlapping peptides, and manual review may be used to improve confidence.
The main advantage is independence from complete reference databases. De novo sequencing can help with non-model species, novel peptides, engineered proteins, natural products, proprietary constructs, or samples with incomplete annotation. It can also support sequence recovery when the expected protein differs from public database entries.
The limitation is uncertainty. De novo interpretation can be affected by similar residue masses, weak spectra, missing fragments, modifications, and incomplete overlap. Leucine and isoleucine are a common example because they share the same nominal mass in MS/MS interpretation. A responsible report should distinguish confident residues, ambiguous positions, and regions requiring validation.
Use de novo protein sequencing when no reliable reference exists or when database search alone cannot answer the question. For critical projects, plan validation through additional digestion, synthetic peptide comparison, recombinant expression, or orthogonal methods.
Peptide Mapping
Peptide mapping is often used when a reference sequence exists and the goal is confirmation. The protein is digested into peptides, LC-MS/MS identifies observed peptides, and the data are mapped to the expected sequence. The output may include coverage, missed regions, expected modifications, sequence variants, and confidence notes.
Peptide mapping is especially useful for recombinant proteins, biologics, biosimilar comparison, and quality-oriented primary-structure work. It can support the statement that observed peptide evidence matches the expected sequence across defined regions.
The limitation is that peptide mapping is not always a discovery tool. If the sequence is unknown, peptide mapping needs a reference to map against. If the reference is wrong, the analysis may miss unexpected regions unless the workflow includes open search, de novo interpretation, or broader protein identification.
Use peptide mapping when the project has a known sequence and needs traceable confirmation. Add de novo analysis or additional methods when unexplained gaps or variants matter.
Comparison Tabl
|
Method
|
Best Fit
|
Main Output
|
Key Limitation
|
| LC-MS/MS protein sequencing | Broad peptide evidence and sequence coverage | Identified peptides, coverage, variants, confidence notes | Coverage may be incomplete |
| Edman sequencing | Accessible N-terminal read | Ordered N-terminal residues | Limited length; blocked N-termini fail |
| N/C terminal sequencing | Protein end processing | Terminal residue evidence | Not full sequence recovery |
| De novo protein sequencing | Unknown or database-limited proteins | Proposed peptide sequences | Ambiguity requires careful reporting |
| Peptide mapping | Known sequence confirmation | Coverage map against reference | Requires a reliable reference |
How to Choose by Project Scenario
For an unknown gel band, start with protein identification or LC-MS/MS protein sequencing. If the database match is strong, the project may not need deeper sequencing. If the organism or sequence is poorly represented, de novo sequencing may be added.
For a recombinant protein with an expected sequence, peptide mapping plus intact mass or terminal checks may be more appropriate than full de novo work. The goal is usually confirmation, not rediscovery.
For a protein with suspected processing, focus on N-terminal or C-terminal sequencing. If the processing site is unknown, combine terminal analysis with LC-MS/MS coverage around the suspected region.
For a non-model species or natural peptide, de novo sequencing should be considered early. The sample may need higher purity, multiple digestion strategies, and more careful interpretation than a routine database-assisted project.
For biologics characterization, method choice should be driven by documentation needs. Peptide mapping, terminal analysis, molecular weight confirmation, and primary-structure analysis may be combined to support a stronger evidence package.

Figure 3. The strongest workflow matches evidence depth to the project scenario.
Vendor Evaluation Criteria
A sequencing provider should ask for sample context before recommending a method. Important details include sample amount, purity, buffer, expected molecular weight, species, known sequence, modifications, terminal processing, and downstream use. A provider that recommends one method for every project may not be evaluating the real risk.
Transparent reporting is also important. The report should explain which regions are supported, which regions are uncertain, and why. Raw spectra, peptide lists, coverage maps, terminal evidence, and method notes can make the result easier to reuse for manuscripts, quality records, or development meetings.
Cost and turnaround should be compared only after scope is clear. A cheaper method may be appropriate for a simple N-terminal check. It may be insufficient for unknown sequence recovery. A longer workflow may be justified if it prevents repeat work or produces evidence that can support the next milestone.
Frequently Asked Questions
1. Which protein sequencing method is most comprehensive?
LC-MS/MS-based workflows are often the most flexible because they can provide broad peptide evidence. However, complete confidence may require terminal analysis, peptide mapping, or de novo interpretation depending on the project.
2. When should Edman sequencing be used?
Use Edman sequencing when a purified protein or peptide has an accessible N-terminus and the project needs a limited direct N-terminal read.
3. Is de novo sequencing always better for unknown proteins?
Not always. De novo sequencing is useful when database information is missing, but it depends on sample quality and strong MS/MS data. Protein identification may be enough when a reliable database match exists.
4. Can peptide mapping replace full sequencing?
Peptide mapping can confirm a known sequence across observed regions. It does not automatically recover unknown regions and should be paired with other methods when unexplained gaps matter.
5. How should researchers compare service providers?
Compare sample review quality, method fit, evidence transparency, reporting detail, raw data access, turnaround assumptions, and willingness to explain uncertainty.
Conclusion
Protein sequencing methods are complementary rather than interchangeable. LC-MS/MS provides broad peptide evidence, Edman sequencing supports accessible N-terminal reads, terminal sequencing answers end-processing questions, de novo sequencing helps when references are weak, and peptide mapping confirms known sequences.
For projects where method choice affects cost, confidence, or timeline, contact MtoZ Biolabs to compare sequencing strategies and select an evidence package matched to the sample and research goal.
How to order?

