





Protein-Small Molecule Interactions Characterization Service
Many drugs target proteins that function as receptors for small-molecule ligands. These ligands bind to and alter the structures of their protein targets, thus modulating their biological activities. Key protein targets in the fields of biology and pharmaceutical research include the nuclear receptors (NRs) that belong to the superfamily of ligand-dependent transcription factors, the G-protein coupled receptors (GPCRs), and protein kinases, which are part of a large family of regulatory proteins. Besides serving as receptors for small molecules, proteins can act directly as drug targets in the form of monoclonal antibodies (mAbs) or soluble decoy receptors that bind and neutralize or alter the functions of host proteins. In every case, the regulation of protein structure and dynamics impacts their functional roles, offering avenues for pharmacological intervention. Consequently, methods to characterize the structural and dynamic aspects of protein-ligand and protein-target complexes are of considerable interest. Atomic structures determined by X-ray crystallography have been pivotal in elucidating the structure-function relationships in proteins and have had a profound impact on both biology and drug discovery. Despite the undeniable benefits of X-ray crystallography, it often fails to reveal details about protein dynamics, providing only limited insights. Detecting long-distance allosteric communications across protein-protein interaction surfaces is also challenging from static crystal structures. Unfortunately, some protein states, such as those without bound ligands or in their apolipoprotein forms, resist crystallization. In the last two decades, hydrogen/deuterium exchange (HDX) combined with mass spectrometry (MS) has emerged as a valuable complement to X-ray crystallography in studying protein structures and dynamics.
HDX is an acid-base catalyzed reaction that labels proteins to report on the local environment of amide hydrogens. This technique, when paired with high-resolution MS, facilitates precise and sensitive data collection (under 1ug per experiment at low μM concentrations) and can detect large proteins and complexes. Performing differential HDX-MS analysis under varying conditions has become an essential tool to assess the impact of chemical modifications, mutations, and binding events on protein stability and conformational dynamics. The development of automated HDX platforms equipped with advanced software has greatly enhanced the speed of data collection and enabled almost real-time data processing and statistical analysis, marking a significant advancement in integrating HDX-MS into drug discovery programs. Linking the deuterium incorporation patterns of various small molecule ligands with functional assays has proven effective in developing structure-activity relationships and defining the functional selectivity among closely related compounds. HDX-MS also provides a means to identify isomeric small molecule binding sites, challenging to locate but crucial for developing more selectively targeted formulations.
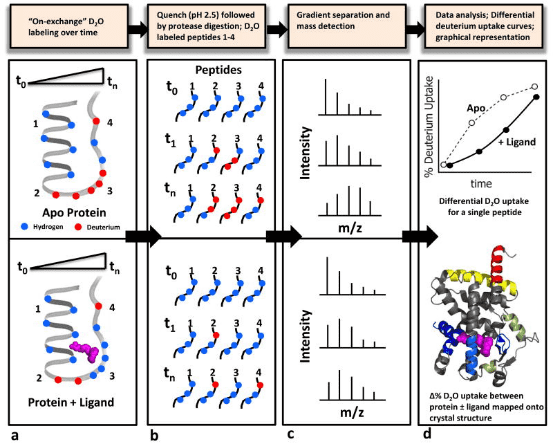
Figure 1. Schematic Diagram of a Typical HDX-MS Workflow [4]
HDX technology is particularly well-suited for characterizing the interactions between protein targets and small molecules that bind to them. A typical HDX ligand screening application involves multiple differential HDX MS experiments to generate a 'HDX fingerprint' for each ligand. Variations among these fingerprints allow for the clustering of ligands into groups with similar binding patterns. It is important to note that the differences in HDX between ligand-free (apo) and ligand-bound (holo) protein states can stem from direct interactions within the ligand binding pocket (LBP) as well as from changes in protein structure or dynamics at regions distant from the binding site. HDX-MS is particularly advantageous over other biophysical methods for conformational analysis as it requires only minimal sample amounts (several hundred pmoL), is highly tolerant of buffer systems, and allows for analysis of samples diluted in solution (above 10 μM). Moreover, HDX-MS is exquisitely sensitive to detecting and quantifying the extent and location of conformational dynamics differences between different protein states. The use of HDX-MS in the entire process of (bio)pharmaceutical drug discovery and development has been expanding, including in the analysis of (a) drug-protein interactions, (b) the conformational properties and stability of protein drugs, and (c) the comparability and consistency among batches of related protein therapeutics.
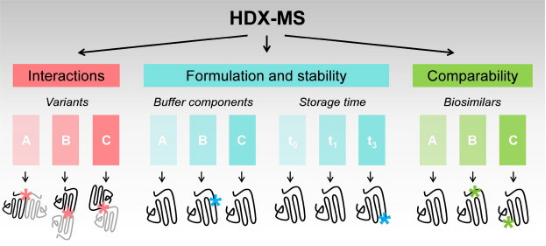
Figure 2. Applications of HDX-MS in Biopharmaceutical Drug Development [5]
The aim of HDX is to explore the structure and dynamics of proteins by substituting the hydrogen atoms covalently bonded in the protein backbone with deuterium atoms. In proteins, hydrogen atoms can be replaced by solvent molecules. There are three types of hydrogen atoms in proteins: Type I, which are bonded to hetero atoms undergoing a fast reversible exchange under all buffer conditions; Type II, which are bonded to amide of the protein backbone exhibiting solvent dependent behaviour (low pH and temperature conditions slow down the exchange rate and these conditions are widely used in HDX experiments); and Type III, which are aliphatic and aromatic hydrogen atoms that typically do not participate in exchange during standard HDX-MS experiments.
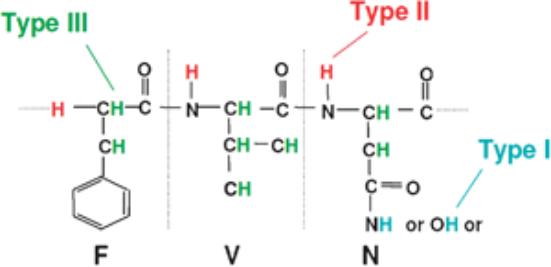
Figure 3. Three Types of Hydrogen Atoms in Proteins [6]
In identifying interactions between proteins and small molecule ligands, the general procedure involves incubating the target protein (± ligand) at pre-determined time points. At the end of the HDX period, the reaction is quenched by adding a cold acidic solution, followed by enzymatic digestion of the protein. Liquid chromatography-electrospray ionization MS (LC-ESI-MS) is used to measure the percentage of deuterium incorporated into each peptide at each time point, plotting the deuterium uptake curve for the "apo" sample, or comparing it with that of the ligand. Deuterium uptake information is typically displayed using a color gradient beneath the protein sequence or superimposed onto a 3D crystal structure image. Differences in deuterium uptake can be visualized in this manner, allowing for the comparison of different ligands.
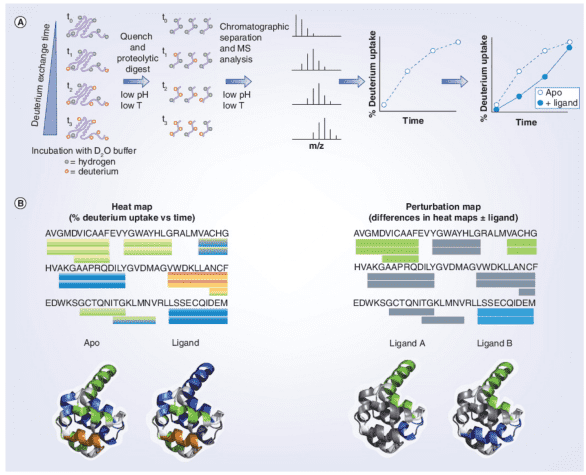
Figure 4. HDX-MS Workflow in Protein-Ligand Interactions [7]
HDX-MS experiments can be conducted in various ways depending on the desired information. Researchers have developed a technique called PLIMSTEX (protein-ligand interaction by MS, titration, and HDX) for monitoring protein-ligand interactions. PLIMSTEX experiments assess the binding affinity of protein-ligand complexes. The target protein is equilibrated with increasing concentrations of the ligand to form complexes at various protein/ligand ratios. Each sample is then diluted into a tritiated buffer and undergoes HDX for a designated period. Samples are purified online via reverse-phase chromatography (RPC), and the MS measures the deuterium uptake. The observed mass difference due to deuterium incorporation decreases as ligand concentration increases, reflecting the protective effect on the backbone amides after ligand binding. The curve relating the protein-ligand ratio to deuterium uptake is determined by the binding affinity and stoichiometry. Recently, PLIMSTEX has been employed to determine the self-association equilibrium constants of proteins, a method now known as SIMSTEX (self-interaction monitoring by MS, titration, and HDX), which has been used to study insulin analogs and may become an essential tool for investigating other large protein self-assemblies.
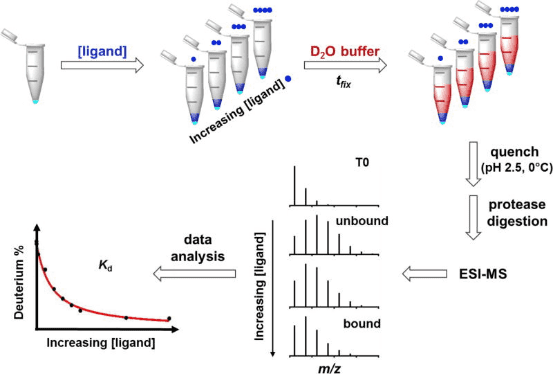
Figure 5. Schematic Diagram of the PLIMSTEX Workflow [8]
Additionally, Fitzgerald and colleagues developed an alternative method using matrix-assisted laser desorption/ionization (MALDI)-MS, known as SUPREX (stability of unpurified proteins from rates of HDX), which can determine the free energy values (ΔGu) related to protein unfolding mechanisms. SUPREX utilizes denaturants, such as guanidine hydrochloride or urea, to assess the thermodynamic stability of proteins. Proteins or protein-ligand complexes are diluted into a series of HDX buffers containing progressively increasing concentrations of chemical denaturants. After incubation, the mass of each denatured protein sample is measured using MALDI-MS. The number of exchanged hydrogens is plotted against the denaturant concentration, indicating that as denaturant concentration increases, more backbone amides are exposed, marking the protein's transition from a folded to an unfolded state. The increased stability of protein-ligand complexes necessitates higher denaturant concentrations to induce their unfolding. By incorporating a proteolytic digestion step, the application scope of the SUPREX approach can be expanded to include large multimeric protein systems.
In the PLIMSTEX analysis (A), the plot shows the relationship between the mass increase (Δm) caused by deuterium uptake and the total ligand concentration; as the ligand concentration increases, the number of protein-ligand complexes increases, thus reducing deuterium uptake. In the SUPREX analysis (B), the plot shows the relationship between Δm and the denaturant concentration. The mass increase is due to the increased deuterium uptake as the protein unfolds; compared to the apolipoprotein (solid line), the maximum deuterium uptake of protein-ligand complexes (dashed line) requires higher concentrations of denaturants.
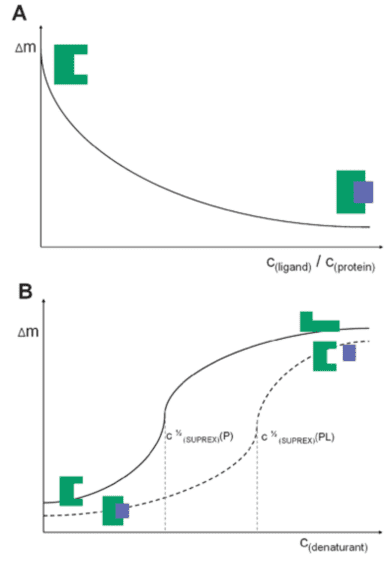
Figure 6. PLIMSTEX (A) and SUPREX (B) Curve Diagrams [8]
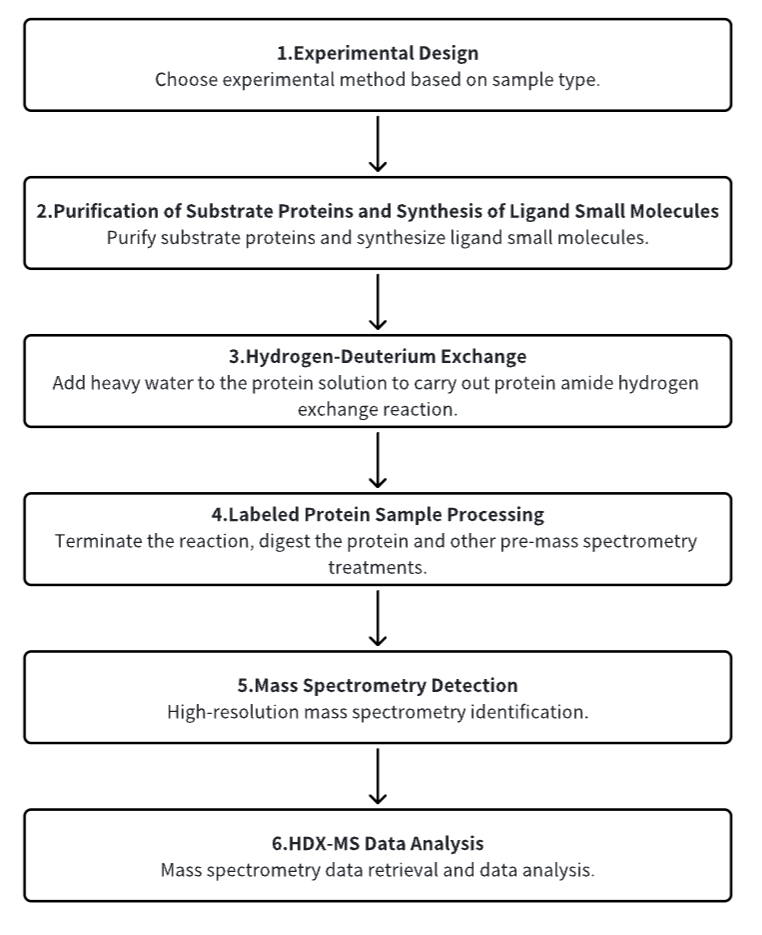
Analysis Workflow
1. Establish the Experimental Workflow Based on Specific Research Needs (Top-Down or Bottom-Up)
2. Purify Substrate Proteins and Synthesize Ligand Small Molecules
3. Hydrogen-Deuterium Exchange
4. Terminate Labeling and Process Protein Samples
5. Mass Spectrometry Detection
6. HDX-MS Data Analysis
Service Advantages
1. Experimental Methods Tailored to Specific Research Requirements
2. Automated Sample Handling Platform for Precise Control of Hydrogen-Deuterium Exchange Conditions
3. High-Resolution and Reliable Mass Spectrometry Analysis
4. Diverse Data Analysis Methods
Sample Results
1. Conformational Elucidation Through Small Molecule Biocoupling and HDX-MS: A Case Study with Human Cytochrome P450 3A4
Researchers have developed a novel method for studying isomers by integrating carefully selected bioconjugates with HDX-MS. This approach avoids issues associated with weak substrate binding and ligand repositioning. The method's efficacy was demonstrated using human cytochrome P450 3A4 (CYP3A4), a critical enzyme in drug metabolism. Drugs' ectopic activation and inhibition of CYP3A4 represent key mechanisms in drug interactions. Our analysis of several CYP3A4-effector bioconjugates revealed that some could simulate the isomeric effects of positive effectors, enhancing activity even when the label did not occupy the isomeric pocket (agonistic) or showed no activation while blocking the isomeric site (antagonistic). This analysis enabled the precise identification of isomeric sites, the related protein structural dynamics, and the presence of coexisting conformations.
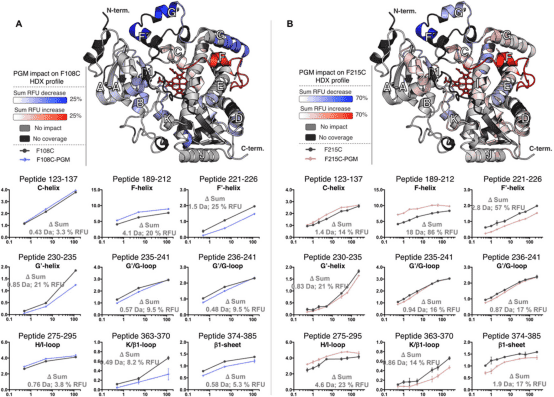
Figure 7. Differential HDX Spectra of PGM Bioconjugates Mimicking Isomers [9]
2. HDX-MS Has Demonstrated Both Orthosteric and Allosteric Changes in the Structural Dynamics of Apolipoprotein-D upon Progesterone Binding
Apolipoprotein-D, a glycosylated tetrameric lipocalin, binds and transports hydrophobic molecules like progesterone and arachidonic acid. Structurally, it features an eight-stranded β-barrel fold stabilized by two intramolecular disulfide bonds and an adjacent α-helix. Crystallography studies of recombinant apolipoprotein‐D demonstrated no major conformational changes upon progesterone binding. HDX-MS, by monitoring amide hydrogen to deuterium exchange, reveals the solution-based structural changes of proteins, often invisible in X-ray crystallography. This technique has proven invaluable in detecting conformational and dynamic shifts triggered by functions like ligand binding. An HDX-MS study tailored for apolipoprotein-D tackled the challenges of the protein's rigidity and low pepsin digestibility through stringent quenching and extended deuteration, yielding 85% sequence coverage and 50% deuterium exchange. This detailed analysis showed that progesterone binding significantly altered deuterium exchange across specific peptide segments, underscoring the dynamic stability imparted by orthosteric effects in the ligand binding pocket combined with allosteric effect at the N‐ and C‐termini. This study provides profound insights into the structural dynamics of apolipoprotein-D and illustrates the impact of progesterone binding on the protein's distal regions.
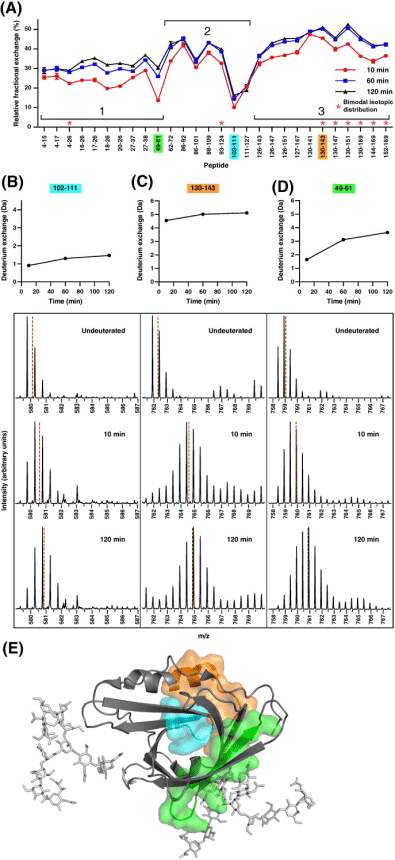
Figure 8. Relative Fraction Deuterium Exchange of Apo-Form of apoD, Highlighting Differences in Exchange and Dynamics [10]
3. HDX-MS Reveals Structural Determinants for RORγ Hyperactivation by Synthetic Agonists
Members of the NR superfamily regulate crucial physiological and pathophysiological processes, ranging from development and metabolism to inflammation and cancer. Targeting these receptors with synthetic small molecules serves both therapeutic purposes to correct abnormal NR signaling and as probes to explore physiological roles. However, nearly half of NRs do not have specific cognate ligands or its unclear if they possess ligand dependent activities and these receptors are called orphans. Studies have shown that the ligand-dependent activity of orphan RORγ can be elucidated by selectively disrupting binding with putative endogenous (not synthetic) ligands. Moreover, characterizing a library of RORγ modulators through HDX-MS linked the receptor's structural dynamics with its functional activity, as validated in biochemical and cell-based assays. These findings are corroborated with X-ray co-crystallography and site-directed mutagenesis to collectively reveal the structural determinants of RORγ ligand-dependent activation, critical for designing full agonists for application in cancer immunotherapy.
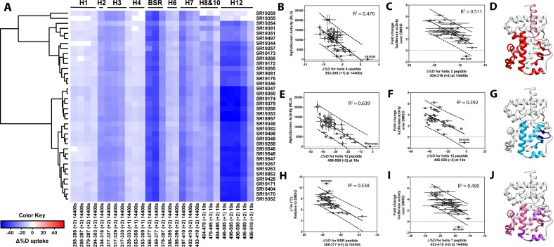
Figure 9. Differential HDX-MS and Activity Screening of 38 RORγ Regulators, Revealing the Structural Determinants of Activation [11]
Sample Submission Requirements
1. Comprehensive Experimental Steps
2. Specifications of Relevant Instrumentation
3. Compilation of Original Experimental Data
4. Data Analysis Report (Protein Spatial Epitope, Conformation, etc. Analysis Results)
5. Report on the Physicochemical Properties and Efficacy of PROTACs
Applications
1. HDX in Small Molecule Screening
HDX technology is ideally suited for screening chemical libraries that include small molecules, natural products, or fragments. The goal is to find chemical entities that produce a specific HDX fingerprint on the target protein. Although using HDX as a screening tool presents challenges, particularly in its application to high-throughput environments capable of screening thousands of compounds, recent studies have proposed promising methods. For example, distinguishing the desired HDX behavior from that of a ligand-free protein at a single time point—such as with the receptor PPARγ—could allow for the screening of 10-20 compounds within several hours. This technique has been validated with PPAR regulators known for their HDX profiles and used to generate initial HDX fingerprints for numerous assumed endogenous ligands of the receptor. The primary limitation in throughput arises from the need for extensive chromatographic steps to separate target peptides from interfering ones. Employing the described multiple reaction monitoring (MRM) technique can accelerate these chromatographic steps and streamline the data analysis process.
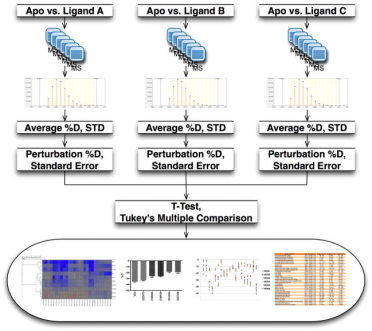
Figure 10. Schematic Diagram of the Differential HDX-MS Screening Workflow for Small Molecule Compounds [12]
FAQ
Q1: What are the applications of HDX-MS in pharmaceutical research?
(1) Ligand Interaction and Epitope Mapping: HDX-MS is valuable for characterizing how ligands interact with their targets and for mapping the sites of interaction (epitopes). This information is crucial for a deeper understanding of drug target engagement and can open new avenues in drug development.
(2) Quality Control and Comparability Analysis: The effectiveness of protein drugs is intimately connected to their conformation, which can be altered by sequence variations and changes arising from protein expression, purification, formulation, or storage. HDX-MS serves as an essential tool for performing quality control and ensuring the comparability of protein drugs.
(3) Protein Conformation Detection: Modifications such as mutations, glycan engineering, and chemical changes can influence the function and stability of protein drugs. HDX-MS allows for the detection and characterization of how these modifications affect protein conformation.
(4) Determining the Impact of Formulations: The formulation of biopharmaceuticals significantly impacts their stability post-storage. Factors like salt concentration, pH, and protein concentration influence the chemical and physical stability of protein drugs. HDX-MS facilitates the comparative analysis of deuterium uptake kinetics, providing insights into structural changes under various conditions, including different storage conditions and buffer solutions.
References
[1] James EI, Murphree TA, Vorauer C, Engen JR, Guttman M. Advances in Hydrogen/Deuterium Exchange Mass Spectrometry and the Pursuit of Challenging Biological Systems. Chem Rev. 2022 Apr 27;122(8):7562-7623. doi: 10.1021/acs.chemrev.1c00279. Epub 2021 Sep 7. PMID: 34493042; PMCID: PMC9053315.
[2] Masson GR, Jenkins ML, Burke JE. An overview of hydrogen deuterium exchange mass spectrometry (HDX-MS) in drug discovery. Expert Opin Drug Discov. 2017 Oct;12(10):981-994. doi: 10.1080/17460441.2017.1363734. Epub 2017 Aug 17. PMID: 28770632.
[3] Damont A, Legrand A, Cao C, Fenaille F, Tabet JC. Hydrogen/deuterium exchange mass spectrometry in the world of small molecules. Mass Spectrom Rev. 2023 Jul-Aug;42(4):1300-1331. doi: 10.1002/mas.21765. Epub 2021 Dec 2. PMID: 34859466.
[4] Marciano DP, Dharmarajan V, Griffin PR. HDX-MS guided drug discovery: small molecules and biopharmaceuticals. Curr Opin Struct Biol. 2014 Oct;28:105-11. doi: 10.1016/j.sbi.2014.08.007. Epub 2014 Aug 30. PMID: 25179005; PMCID: PMC4253076.
[5] Leurs U, Mistarz UH, Rand KD. Getting to the core of protein pharmaceuticals--Comprehensive structure analysis by mass spectrometry. Eur J Pharm Biopharm. 2015 Jun;93:95-109. doi: 10.1016/j.ejpb.2015.03.012. Epub 2015 Mar 17. PMID: 25791210.
[6] Pacholarz KJ, Garlish RA, Taylor RJ, Barran PE. Mass spectrometry based tools to investigate protein-ligand interactions for drug discovery. Chem Soc Rev. 2012 Jun 7;41(11):4335-55. doi: 10.1039/c2cs35035a. Epub 2012 Apr 24. PMID: 22532017.
[7] Michael J Chalmers, Scott A Busby, Bruce D Pascal, Graham M West & Patrick R Griffin (2011) Differential hydrogen/deuterium exchange mass spectrometry analysis of protein–ligand interactions, Expert Review of Proteomics, 8:1, 43-59, DOI: 10.1586/epr.10.109
[8] Pacholarz KJ, Garlish RA, Taylor RJ, Barran PE. Mass spectrometry based tools to investigate protein-ligand interactions for drug discovery. Chem Soc Rev. 2012 Jun 7;41(11):4335-55. doi: 10.1039/c2cs35035a. Epub 2012 Apr 24. PMID: 22532017.
[9] Ducharme J, Polic V, Thibodeaux CJ, Auclair K. Combining Small-Molecule Bioconjugation and Hydrogen-Deuterium Exchange Mass Spectrometry (HDX-MS) to Expose Allostery: the Case of Human Cytochrome P450 3A4. ACS Chem Biol. 2021 May 21;16(5):882-890. doi: 10.1021/acschembio.1c00084. Epub 2021 Apr 29. PMID: 33913317.
[10] Kielkopf CS, Ghosh M, Anand GS, Brown SHJ. HDX-MS reveals orthosteric and allosteric changes in apolipoprotein-D structural dynamics upon binding of progesterone. Protein Sci. 2019 Feb;28(2):365-374. doi: 10.1002/pro.3534. Epub 2018 Dec 20. PMID: 30353968; PMCID: PMC6319766.
[11] Strutzenberg TS, Garcia-Ordonez RD, Novick SJ, Park H, Chang MR, Doebellin C, He Y, Patouret R, Kamenecka TM, Griffin PR. HDX-MS reveals structural determinants for RORγ hyperactivation by synthetic agonists. Elife. 2019 Jun 7;8:e47172. doi: 10.7554/eLife.47172. Erratum in: Elife. 2019 Oct 18;8: PMID: 31172947; PMCID: PMC6579513.
[12] Chalmers MJ, Pascal BD, Willis S, Zhang J, Iturria SJ, Dodge JA, Griffin PR. Methods for the Analysis of High Precision Differential Hydrogen Deuterium Exchange Data. Int J Mass Spectrom. 2011 Apr 30;302(1-3):59-68. doi: 10.1016/j.ijms.2010.08.002. PMID: 21528013; PMCID: PMC3081588.
MtoZ Biolabs, an integrated chromatography and mass spectrometry (MS) services provider.
Related Services
Protein-Protein Interactions Characterization Service | HDX-MS
Antibody-Antigen Interactions Characterization Service | HDX-MS
How to order?

