





Protein-Protein Interactions Characterization Service | HDX-MS
All cellular protein functions, including those involved in disease processes, require coordinated movements at the atomic or domain level, known as conformational dynamics. Proteins regulate almost all biological processes within living organisms and are the material basis of life activities. Proteins fold into specific three-dimensional structures based on their primary structure (amino acid sequence), and their functions are regulated by structural dynamics, interactions with ligands (such as small molecules, proteins, or DNA), and post-translational modifications (PTMs). Thus, deepening our understanding of the structure and function of biomacromolecules not only sheds light on the essence of biological phenomena but also has profound implications for human health and significantly advances drug target discovery and new drug development.
Understanding protein dynamics is essential for drug development and identifying the potential molecular bases of many diseases. HDX-MS provides invaluable insights into protein dynamics, complementing the static images offered by traditional high-resolution structural tools such as X-ray crystallography and structural nuclear magnetic resonance (NMR). HDX-MS requires only a fraction of the protein quantity needed for other biophysical techniques, which typically offer lower resolution. The use of HDX-MS in both industrial and academic settings is rapidly increasing and has proven successful in numerous drug and vaccine development endeavors, playing a critical role in understanding conformational effects and mapping binding sites.
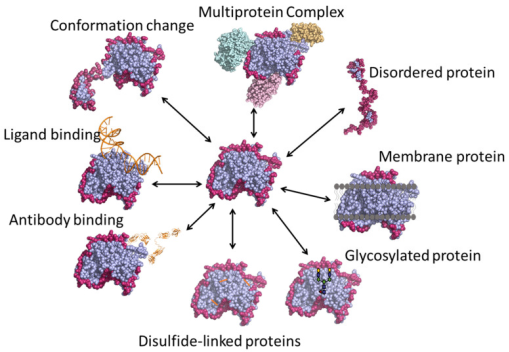
Figure 1. Various Applications of HDX-MS Technology [4]
Protein-protein interactions (PPIs) participate in numerous intracellular physiological processes, such as signal transduction, molecular transport, and various metabolic pathways. Dysfunctional PPIs are causal factors in many diseases, making the identification of PPI interfaces within organisms crucial for drug design and development. Researchers have developed multiple methods to study PPIs, including MS-based techniques such as affinity capture-MS, proximity label-MS, and co-fractionation-MS, which together contribute over 60% of the data in BioGRID, one of the largest protein interaction databases.
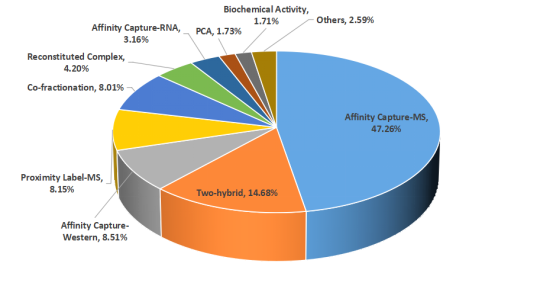
Figure 2. Experimental Methods for Detecting Protein Interactions
Epitopes are typically classified into two types: linear and conformational. Linear epitopes consist of consecutive amino acid sequences (8-15 residues) known as continuous epitopes. Conformational epitopes, on the other hand, are made up of discrete fragments that, while separate in the primary sequence, come together in the three-dimensional structure and are known as discontinuous epitopes. Most antibody-antigen interactions, especially those involving autoantibodies and protective antibodies (such as those from vaccines), are determined by conformational epitopes. The specific amino acid sequences of the contact areas in most antigen-antibody interactions are often unknown. Breakthroughs in experimental techniques are urgently needed to rapidly identify the interacting interfaces of antigens/antibodies in solution. Recently, various experimental and theoretical methods for epitope mapping have been introduced. Epitope mapping techniques are categorized into immunochemical analysis, structural analysis, and computational analysis.
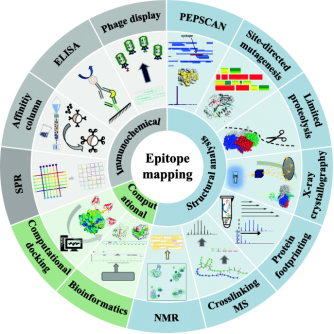
Figure 3. Strategies for Epitope Mapping Analysis [5]
In HDX technology, amide hydrogen atoms in the protein backbone exchange with deuterium atoms (heavy hydrogen) in the environment. This technique employs deuterium atoms as structural probes to gather information about protein structures. By monitoring changes in deuterium incorporation over time, detailed data on the protein's structure, dynamics, and interactions can be obtained. HDX-MS offers many advantages for epitope mapping, such as high sensitivity, low sample consumption, rapid analysis, high throughput, and no upper limit on molecular weight. Moreover, the labeling reaction can proceed under nearly all physiological conditions, ensuring maximal preservation of the protein's native active state throughout the experimental process.
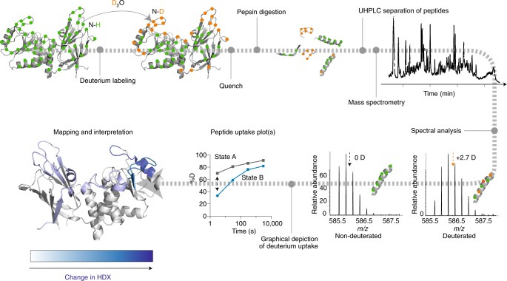
Figure 4. HDX-MS Analysis Workflow [6]
The backbone amide hydrogens of proteins exchange with deuterium nuclei in the solution. This exchange rate is influenced by factors such as pH, temperature, hydrogen bonding, and solvent accessibility. With proper control of pH and temperature, deuteration reports on conformation and conformational changes affected by hydrogen bonding and solvent accessibility. Typically, poorly hydrogen bonded and/or solvent exposed regions will incorporate deuterium more quickly than those that are strongly hydrogen bonded and/or solvent protected. A common method involves comparing different conformational states of the same protein and documenting the exchange and conformational differences between these states. In a typical multiprotein HDX-MS experiment, each protein, whether in isolation or in complex, follows the same workflow: labeling with deuterium at physiological pH, quenching of the labeling reaction, digestion, MS measurement of deuterium, and data processing.
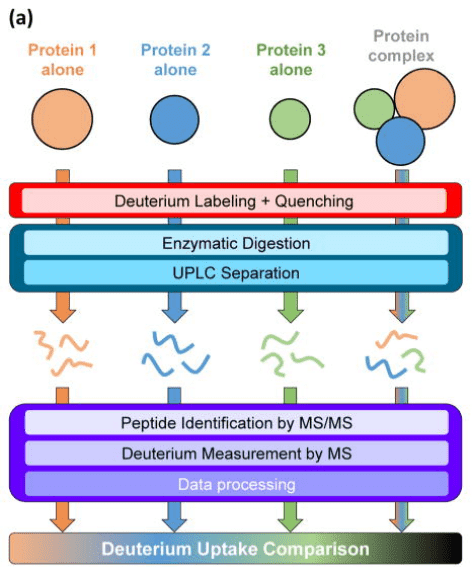
Figure 5. HDX-MS Analysis Process for Protein Interactions [7]
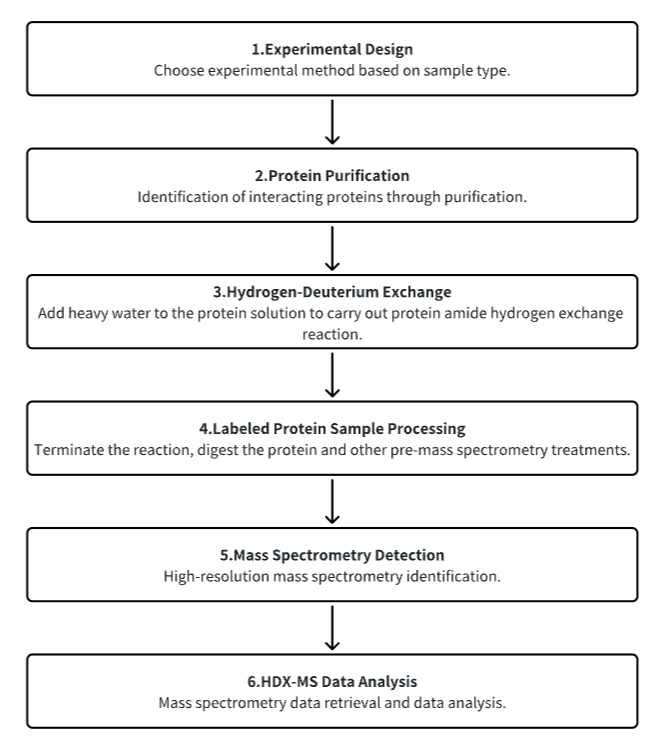
Analysis Workflow
1. Establish the Experimental Workflow Based on Specific Research Needs (Top-Down or Bottom-Up)
2. Purify Protein
3. Hydrogen-Deuterium Exchange
4. Terminate Labeling and Process Protein Samples
5. Mass Spectrometry Detection
6. HDX-MS Data Analysis
Service Advantages
1. Experimental Methods Tailored to Specific Research Requirements
2. Automated Sample Handling Platform for Precise Control of Hydrogen-Deuterium Exchange Conditions
3. High-Resolution and Reliable Mass Spectrometry Analysis
4. Diverse Data Analysis Methods
Sample Results
1. Application of HDX-MS in Chaperones and Chaperone-Assisted Protein Folding
HDX-MS is an ideal method for monitoring protein folding and unfolding. The exchange of deuterium in solution with amide hydrogens in the protein varies significantly based on the degree of folding and the protection of the backbone amide positions. Molecular chaperones, essential for protein folding in cells, can be deeply understood through HDX-MS, which elucidates their role and mechanisms during the protein folding process. Although HDX-MS has long been desired for studying protein folding issues, technical challenges have hindered many studies. Over the past 20 years, research has been conducted on the conformational changes of chaperones themselves (such as GroEL/GroES, Hsp70, and Hsp90). Studies on the interactions between chaperones, co-chaperones, and substrate proteins have revealed binding interfaces, allosteric conformational changes, and component remodeling during various chaperone cycles. Experiments have also clarified how chaperones facilitate and enhance the folding pathways of substrate proteins.
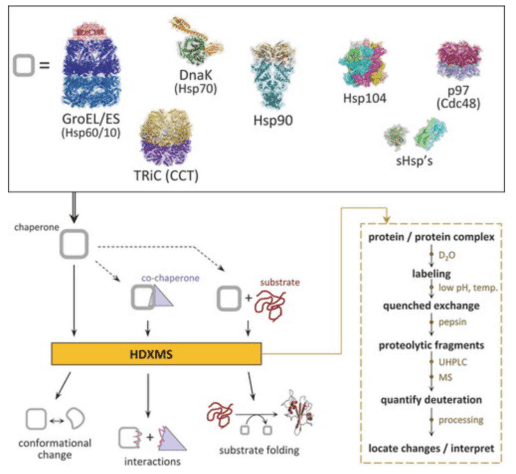
Figure 6. HDX-MS Analysis Workflow for Protein Folding [8]
2. TMEM106B as a Receptor Mediating ACE2-Independent SARS-CoV-2 Cell Entry
SARS-CoV-2 is associated with broad tissue tropism, a characteristic often determined by the availability of entry receptors on host cells. Studies have identified the lysosomal transmembrane protein TMEM106B as an alternative receptor for SARS-CoV-2 entry into angiotensin-converting enzyme (ACE2)-negative cells. The spike substitution E484D enhances binding to TMEM106B, thereby boosting TMEM106B-mediated entry. TMEM106B-specific monoclonal antibodies block SARS-CoV-2 infection, confirming the role of TMEM106B in viral entry. Utilizing X-ray crystallography, cryogenic electron microscopy (cryo-EM), and HDX-MS, it was discovered that the lumenal domain (LD) of TMEM106B engages with the receptor-binding motif of the SARS-CoV-2 spike. Ultimately, it was found that TMEM106B promotes spike-mediated syncytium formation, indicating its role in viral fusion. These findings identify an ACE2-independent SARS-CoV-2 infection mechanism that involves cooperative interactions with the receptors heparan sulfate and TMEM106B.
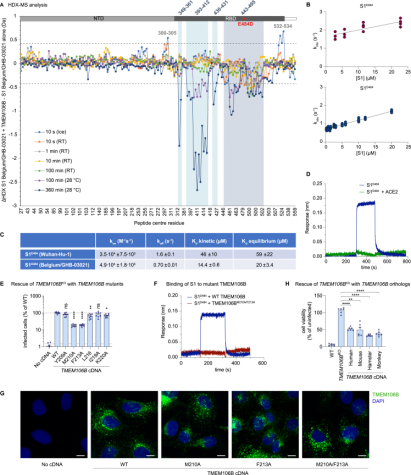
Figure 7. HDX-MS Confirms RBD-TMEM106B Interaction [9]
3. Ligand-Induced Conformational Interactions between Pseudomonas Aeruginosa Heme Binding Protein and Heme Oxygenase
The C-terminal structural domain of the heme binding protein (PhuS) requires heme-dependent conformational rearrangement to interact with the iron-regulated heme oxygenase (HemO). Studies employing site-directed mutagenesis, resonance Raman (RR), HDX-MS, and molecular dynamics (MD) have further explored the fundamental mechanisms of this conformational rearrangement and its effects on heme transfer. HDX-MS revealed that the apo-PhuS C-terminal α6/α7/α8-helices are largely unstructured, while the apo-PhuS H212R variant showed increased structuring in these regions. Compared to the wild type (WT), the apo-PhuS H212R showed increased heme binding and did not detect the five-coordinate high-spin (5cHS) heme intermediate, consistent with a more folded C-terminus and reduced dynamics. HDX-MS and MD of holo-PhuS showed an overall reduction in molecular flexibility of the entire protein and significant structural rearrangement and protection of the heme-binding pocket. Slow cooperative unfolding/folding events were observed in the C-terminal helices and N-terminal α1/α2-helices of holo-PhuS, which were suppressed or eliminated in the holo-PhuS H212R variant. Chemical cross-linking and MALDI-TOF MS mapped these same regions to the PhuS: HemO protein-protein interface. This study provides significant insights into the role of protein dynamics in the binding and release of heme within bacterial heme transport proteins.
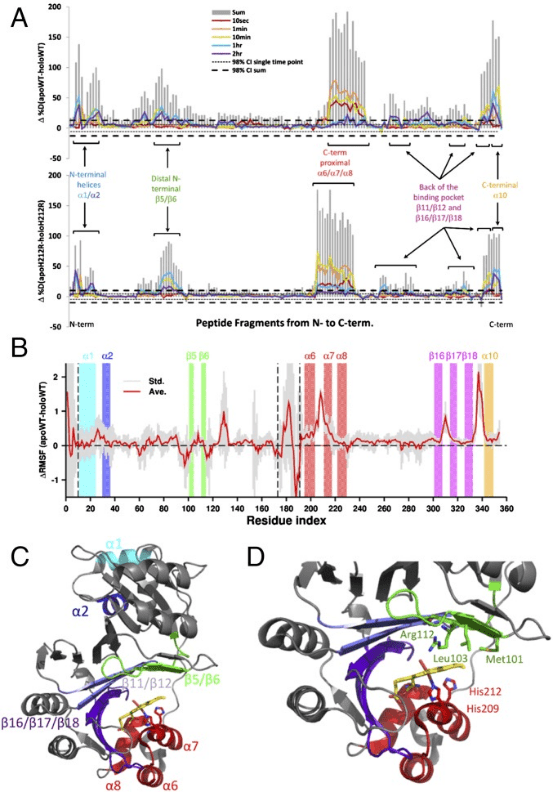
Figure 8. HDX-MS Detection of PhuS's Effects on Heme Transfer [10]
Sample Submission Requirements
1. Comprehensive Experimental Steps
2. Specifications of Relevant Instrumentation
3. Compilation of Original Experimental Data
4. Data analysis Report (Protein Spatial Epitope, Conformation, etc. Analysis Results)
Applications
1. Using HDX-MS to Explore Protein-Membrane Interactions and Dynamics
The cell membrane serves as a central hub for the initiation and execution of many signaling processes. Crucial to these processes is the highly controlled recruitment and assembly of proteins on the membrane surface. HDX-MS facilitates the study of the molecular mechanisms mediating protein-membrane interactions. HDX-MS is a powerful analytical technique that allows for the measurement of the exchange rate of backbone amide hydrogens with solvent to make inferences about protein structure and conformation.
2. HDX-MS Studies on Intrinsically Disordered Proteins
Intrinsically disordered proteins (IDPs) lack a unique 3D structure and adopt a wide range of highly dynamic conformations in solution. Although IDPs are disordered in their native structures, they can form localized transient secondary structures, referred to as residual structures. IDPs or intrinsically disordered regions (IDRs) in proteins play crucial roles in many biological processes, such as cell signaling, gene regulation, and PTMs. The highly dynamic nature of IDPs complicates protein crystallization, making NMR and HDX-MS valuable tools for studying IDPs. However, NMR requires high concentrations of protein samples, which can affect the stability of many proteins. Thus, HDX-MS can serve as a useful tool for studying the conformational dynamics of IDPs.
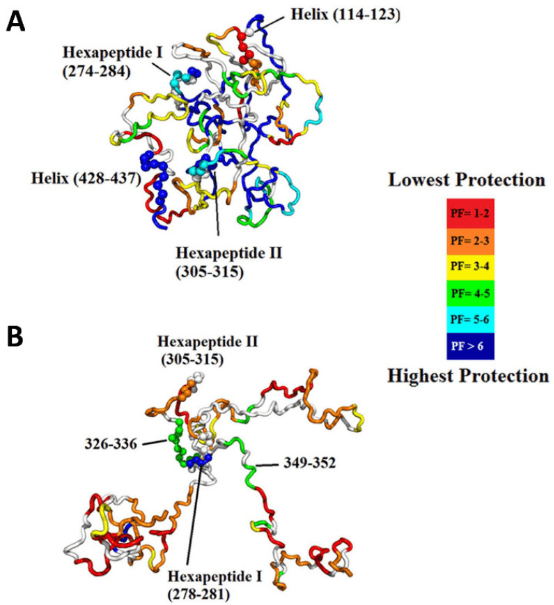
Figure 9. Representative Structures of Native Assemblies [4]
3. HDX-MS Studies on Multi-Protein Complexes
A particular advantage of HDX-MS is the structural characterization of large or multi-protein complexes. Many cellular processes require assembly or interactions between multiple proteins, and the study of large complexes by standard methods, such as X-ray crystallography, and NMR is often prohibitively challenging. With advances in liquid chromatography systems and mass spectrometry resolution, it is now possible to characterize large proteins and protein complexes using a bottom-up approach with HDX-MS.
4. HDX-MS Studies on Glycoproteins
Glycoproteins perform highly diverse functions in cells, ranging from cell-cell recognition and immune functions to pathogen identification. Studying glycoproteins using HDX-MS involves several challenges such as the limited digestion of protein due to steric hindrance from glycans or difficulty in the identification of peptides due to the heterogeneity in the glycan composition. Increasingly, studies are proposing new research strategies, such as labeling glycoproteins in their fully native glycosylated state followed by post-quench steps where glycans are further digested by acid-active endoglycosidases to remove polysaccharides, and simply analyzing the deglycosylated peptides by LC-MS.
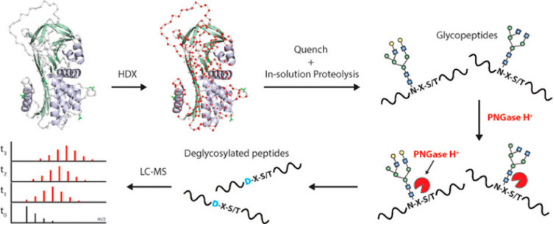
Figure 10. HDX-MS Deglycosylation Steps of Glycoproteins After Quenching [1]
FAQ
Q1: What should be considered during HDX-MS experiments?
References
[1] James EI, Murphree TA, Vorauer C, Engen JR, Guttman M. Advances in Hydrogen/Deuterium Exchange Mass Spectrometry and the Pursuit of Challenging Biological Systems. Chem Rev. 2022 Apr 27;122(8):7562-7623. doi: 10.1021/acs.chemrev.1c00279. Epub 2021 Sep 7. PMID: 34493042; PMCID: PMC9053315.
[2] Masson GR, Jenkins ML, Burke JE. An overview of hydrogen deuterium exchange mass spectrometry (HDX-MS) in drug discovery. Expert Opin Drug Discov. 2017 Oct;12(10):981-994. doi: 10.1080/17460441.2017.1363734. Epub 2017 Aug 17. PMID: 28770632.
[3] Damont A, Legrand A, Cao C, Fenaille F, Tabet JC. Hydrogen/deuterium exchange mass spectrometry in the world of small molecules. Mass Spectrom Rev. 2023 Jul-Aug;42(4):1300-1331. doi: 10.1002/mas.21765. Epub 2021 Dec 2. PMID: 34859466.
[4] Narang D, Lento C, J Wilson D. HDX-MS: An Analytical Tool to Capture Protein Motion in Action. Biomedicines. 2020 Jul 17;8(7):224. doi: 10.3390/biomedicines8070224. PMID: 32709043; PMCID: PMC7399943.
[5] Sun H, Ma L, Wang L, Xiao P, Li H, Zhou M, Song D. Research advances in hydrogen-deuterium exchange mass spectrometry for protein epitope mapping. Anal Bioanal Chem. 2021 Apr;413(9):2345-2359. doi: 10.1007/s00216-020-03091-9. Epub 2021 Jan 6. PMID: 33404742.
[6] Masson GR, Burke JE, Ahn NG, Anand GS, Borchers C, Brier S, Bou-Assaf GM, Engen JR, Englander SW, Faber J, Garlish R, Griffin PR, Gross ML, Guttman M, Hamuro Y, Heck AJR, Houde D, Iacob RE, Jørgensen TJD, Kaltashov IA, Klinman JP, Konermann L, Man P, Mayne L, Pascal BD, Reichmann D, Skehel M, Snijder J, Strutzenberg TS, Underbakke ES, Wagner C, Wales TE, Walters BT, Weis DD, Wilson DJ, Wintrode PL, Zhang Z, Zheng J, Schriemer DC, Rand KD. Recommendations for performing, interpreting and reporting hydrogen deuterium exchange mass spectrometry (HDX-MS) experiments. Nat Methods. 2019 Jul;16(7):595-602. doi: 10.1038/s41592-019-0459-y. Epub 2019 Jun 27. PMID: 31249422; PMCID: PMC6614034.
[7] Harrison RA, Engen JR. Conformational insight into multi-protein signaling assemblies by hydrogen-deuterium exchange mass spectrometry. Curr Opin Struct Biol. 2016 Dec;41:187-193. doi: 10.1016/j.sbi.2016.08.003. Epub 2016 Aug 20. PMID: 27552080; PMCID: PMC5154849.
[8] Georgescauld F, Wales TE, Engen JR. Hydrogen deuterium exchange mass spectrometry applied to chaperones and chaperone-assisted protein folding. Expert Rev Proteomics. 2019 Jul;16(7):613-625. doi: 10.1080/14789450.2019.1633920. Epub 2019 Jun 24. PMID: 31215268.
[9] Baggen J, Jacquemyn M, Persoons L, Vanstreels E, Pye VE, Wrobel AG, Calvaresi V, Martin SR, Roustan C, Cronin NB, Reading E, Thibaut HJ, Vercruysse T, Maes P, De Smet F, Yee A, Nivitchanyong T, Roell M, Franco-Hernandez N, Rhinn H, Mamchak AA, Ah Young-Chapon M, Brown E, Cherepanov P, Daelemans D. TMEM106B is a receptor mediating ACE2-independent SARS-CoV-2 cell entry. Cell. 2023 Aug 3;186(16):3427-3442.e22. doi: 10.1016/j.cell.2023.06.005. Epub 2023 Jul 7. PMID: 37421949; PMCID: PMC10409496.
[10] Deredge DJ, Huang W, Hui C, Matsumura H, Yue Z, Moënne-Loccoz P, Shen J, Wintrode PL, Wilks A. Ligand-induced allostery in the interaction of the Pseudomonas aeruginosa heme binding protein with heme oxygenase. Proc Natl Acad Sci U S A. 2017 Mar 28;114(13):3421-3426. doi: 10.1073/pnas.1606931114. Epub 2017 Mar 13. PMID: 28289188; PMCID: PMC5380046.
MtoZ Biolabs, an integrated chromatography and mass spectrometry (MS) services provider.
Related Services
Antibody-Antigen Interactions Characterization Service | HDX-MS
Protein-Small Molecule Interactions Characterization Service
How to order?

