





PhIP-Seq: Methods, Applications and Challenges
-
Selecting or constructing a phage-displayed peptide library.
-
Preparing serum, plasma, or another antibody-containing sample.
-
Incubating samples with the peptide library under controlled conditions.
-
Capturing antibody-bound phage through immunoprecipitation.
-
Sequencing enriched phage DNA.
-
Mapping sequencing reads to peptide identities.
-
Filtering candidates with controls, statistics, and biological context.
-
Validating selected candidates with independent assays.
-
Sample type, volume, and antibody abundance.
-
Collection tube, processing time, and storage temperature.
-
Freeze-thaw history, hemolysis, lipemia, and contamination.
-
Disease group, control group, clinical metadata, and treatment history.
-
Batch randomization and consistent sample input.
-
Sequencing quality metrics and mapped read rate.
-
Input library representation and peptide coverage.
-
Enrichment scores for each peptide or peptide region.
-
Differential enrichment between sample groups.
-
Heatmaps, volcano plots, clustering maps, or peptide coverage tracks.
-
Candidate ranking with background filtering.
-
Recommended validation assays for selected candidates.
-
Library mismatch — relevant epitopes are absent or poorly represented.
-
Library imbalance — some clones dominate the input before antibody capture.
-
Background binding — peptides bind nonspecifically to beads, phage, or sample components.
-
Weak antibody signal — low-abundance antibodies may fall below detection.
-
Cohort bias — disease and control groups differ in collection, storage, or exposure history.
-
Overinterpretation — candidate peptides are treated as validated biomarkers too early.
Introduction
Antibody-focused research often reaches a difficult point when a sample shows immune reactivity but the recognized antigen is not known. A serum cohort may contain disease-associated antibodies, vaccine-induced signatures, infection-history signals, or autoantibody patterns. Targeted assays can confirm known antigens, but targeted assays are less efficient when the antigen space is broad, heterogeneous, or poorly defined. Researchers need a screening strategy that can compare antibody binding across many peptide sequences while still producing results that can be prioritized for validation.
PhIP-Seq, also called phage immunoprecipitation sequencing, was developed for this type of discovery question. The method combines phage display peptide libraries, antibody capture, next- generation sequencing, and enrichment analysis. PhIP-Seq can help researchers identify antibody-reactive peptides, compare serological profiles across groups, and nominate candidate epitope regions. The method is powerful, but the method is not automatic proof of disease mechanism or diagnostic value. Library design, sample selection, controls, and validation determine whether the data become useful evidence.
For teams planning antibody profiling, epitope discovery, vaccine response analysis, or serological biomarker research, MtoZ Biolabs can help evaluate whether the study design matches the biological question, sample set, and downstream validation plan.
Related Services
| Research Need | Recommended Service |
|
Broad antibody reactivity profiling from serum or plasma |
PhIP-Seq Antibody Analysis Service |
| Peptide-level epitope discovery after screening | Antibody Epitope Mapping Service |
| Targeted validation of candidate peptide regions | Peptide Array-Based Epitope Mapping Service |
| Antibody sequence support for follow-up discovery programs | Antibody Sequencing Service |
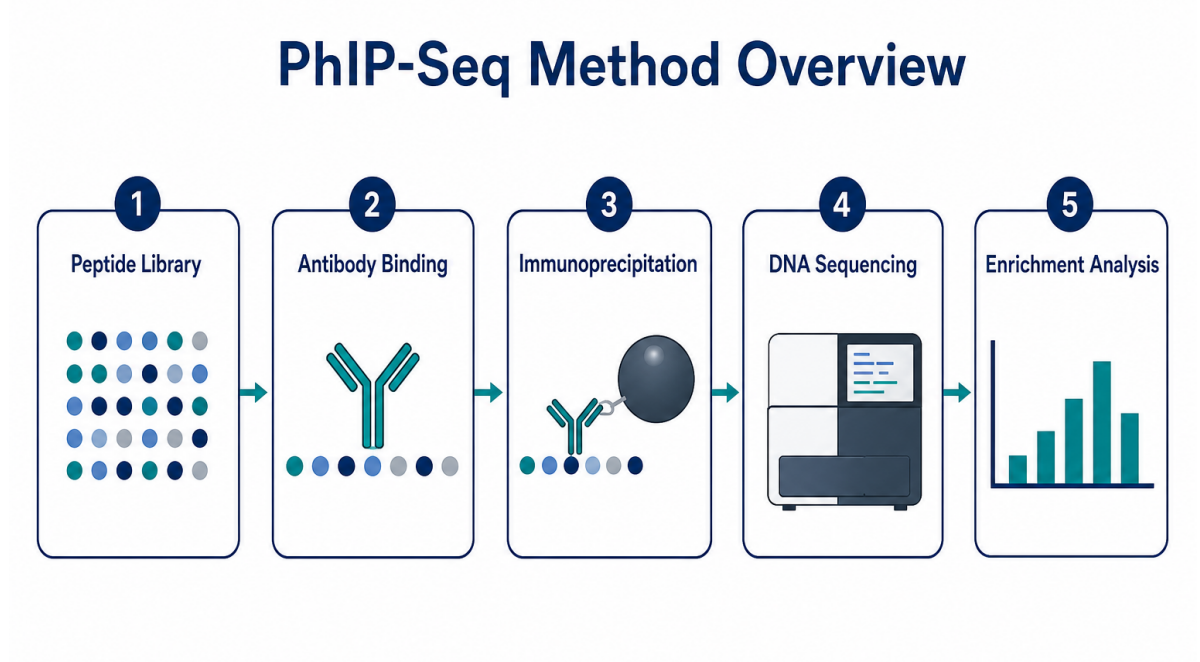
Figure 1. PhIP-Seq links antibody binding to sequencing-based peptide enrichment.
Core Method: Display, Capture, Sequence, Analyze
The central principle of the assay is straightforward. A phage display library presents many peptide sequences on phage particles. Each displayed peptide is linked to a DNA sequence that identifies the peptide. Antibody-containing samples are incubated with the library. Antibodies bind displayed peptides that resemble recognized antigen regions. Immunoprecipitation captures antibody-bound phage particles. Sequencing then identifies which peptide-encoding DNA sequences are enriched.
The resulting dataset is usually a peptide-by-sample matrix. Rows represent peptides, peptide regions, or antigen annotations. Columns represent samples. Enrichment analysis compares post- capture reads with input library reads, negative controls, replicates, or sample groups. Strong interpretation comes from consistent enrichment patterns—not from a single high read count alone.
A typical workflow includes:
Main Method Variants
The assay is not one fixed experimental format. The library type and comparison strategy change the kind of question the experiment can answer. A broad proteome-wide screen can support discovery, while a focused custom library can test a narrower biological hypothesis.
| Method Variant | Best Use Case | Strength | Main Limitation |
| Human proteome library | Autoantibody discovery and disease-associated immune profiling | Broad antigen coverage | Higher background and multiple testing burden |
| Pathogen peptide library | Infection-history studies and vaccine response analysis | Strong fit for exposure- related questions | Only detects represented pathogen peptides |
| Custom tiled library | Focused epitope discovery for selected proteins or variants | Better control over antigen scope | Requires prior target selection |
| Pan-viral or multi- pathogen library | Serological profiling across many infectious agents | Wide immune- history screening | Cross-reactivity may complicate interpretation |
Library design should consider peptide length, tiling density, overlap, clone representation, sequence redundancy, and expected epitope type. Peptide libraries are strongest for linear epitopes and motif-like recognition. Conformational epitopes, glycan-dependent epitopes, lipid antigens, or native protein-complex epitopes often require orthogonal validation methods.
Where the Screening Method Is Used
The screening method is most valuable when researchers need broad antibody profiling rather than single-antigen confirmation. The assay can be used early in discovery to identify candidate regions, or the assay can be used in comparative studies to profile immune recognition across cohorts.
| Application Area | Typical Research Question | Practical Output |
| Autoimmune disease research | Which self-peptides show disease- associated antibody reactivity? | Candidate autoantigens or antibody-reactive regions |
| Infectious disease serology | Which pathogen-derived peptides are enriched in exposed samples? | Exposure-related signatures or immune-history profiles |
| Vaccine response analysis | How does peptide-level recognition change after immunization? | Response breadth, immunodominant regions, group- level profiles |
| Cancer immunology | Which tumor-associated peptides show antibody reactivity? | Candidate tumor-antigen regions for validation |
| Biomarker discovery | Which peptide signals distinguish disease and control groups? | Candidate serological biomarkers for follow-up testing |
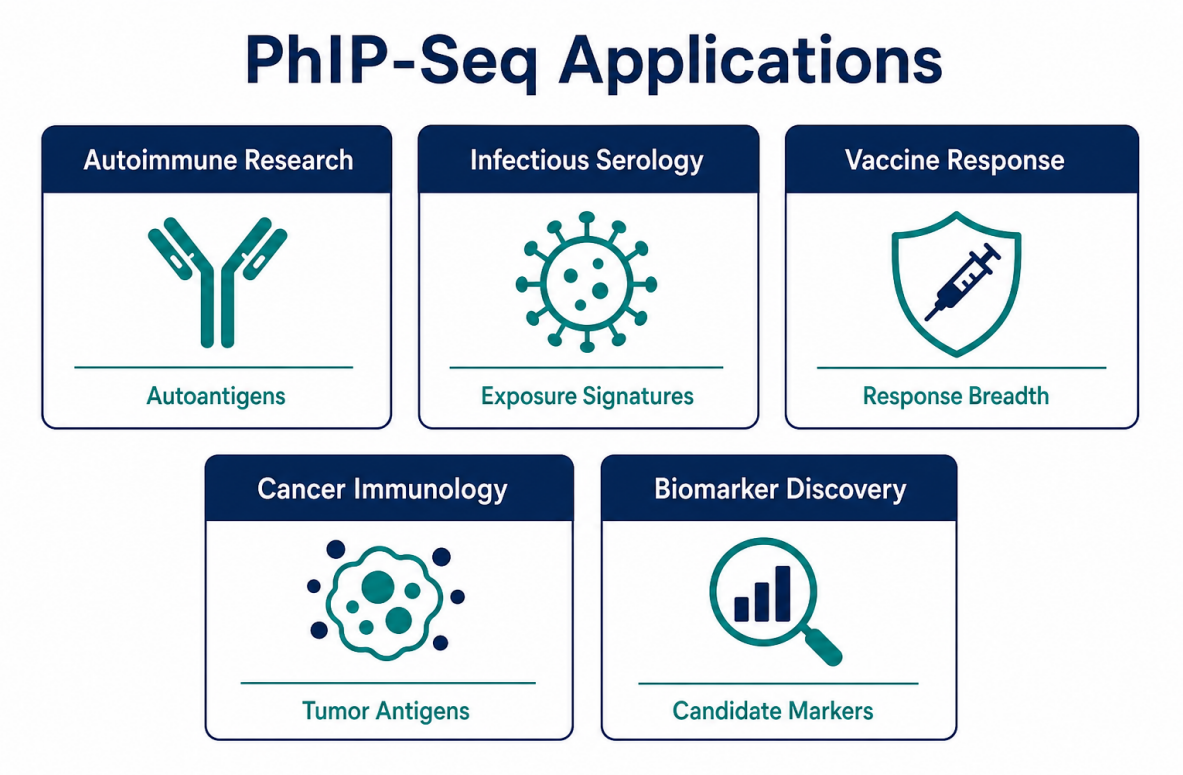
Figure 2. Applications depend on the peptide library, cohort design, and validation goal.
Sample and Control Requirements
Sample quality affects every stage of the workflow. Serum and plasma are common because circulating antibodies are accessible and stable when handled properly. Other antibody- containing fluids may be considered, but sample matrix effects and antibody abundance should be evaluated before full-scale screening.
Practical sample planning should cover:
Controls are essential because phage particles, beads, capture reagents, and sample matrix components can create background. A no-serum control helps estimate nonspecific pull-down. Input library sequencing helps measure starting representation. Technical replicates help evaluate reproducibility. Biological controls help define group-specific enrichment.
| Requirement | Why Requirement Matters | Risk If Missing |
| Input library sequencing | Measures starting clone representation | Library imbalance may be mistaken for enrichment |
| No-serum control | Detects bead or phage background | Nonspecific binders may become false positives |
| Technical replicates | Tests workflow reproducibility | Unstable hits may be overinterpreted |
| Matched biological controls | Supports disease-group comparison | Background exposure may look disease-specific |
| Metadata tracking | Links signals to clinical context | Batch effects may resemble biology |
Data Outputs and Interpretation
A useful report should not stop at a ranked peptide list. The report should connect sequencing reads to peptide counts, enrichment statistics, candidate regions, antigen annotations, and validation recommendations. A peptide can be interesting because of strong enrichment, but a peptide becomes more actionable when neighboring peptides, replicates, controls, and clinical metadata support the same interpretation.
Common output elements include:
For discovery projects, candidate peptides should be treated as leads. Peptide arrays, ELISA, Western blotting, targeted immunoassays, or protein-based assays may be needed before a candidate is used as biomarker evidence or mechanistic evidence.

Figure 3. Strong outputs connect raw reads, enrichment analysis, candidate ranking, and validation planning.
Technical Challenges and How to Manage Them
The main challenge is not generating sequencing reads. The main challenge is generating interpretable enrichment. Several issues can affect interpretation:
The best mitigation strategy is systematic design. Researchers should define the biological question, choose the library accordingly, include controls, randomize processing order, monitor QC metrics, and validate candidates independently. A pilot run can be valuable when samples are limited, the library is new, or the expected signal is uncertain.
| Challenge | Practical Response |
| High background enrichment | Add no-serum controls, improve blocking, review wash stringency |
| Poor replicate agreement | Check library input, sample handling, capture consistency, and sequencing depth |
| Too many candidate hits | Use background filters, group consistency, and antigen-level annotation |
| Weak expected signal | Review sample quality, antibody abundance, serum dilution, and capture conditions |
| Unclear biological meaning | Prioritize candidates with metadata support and plan orthogonal validation |
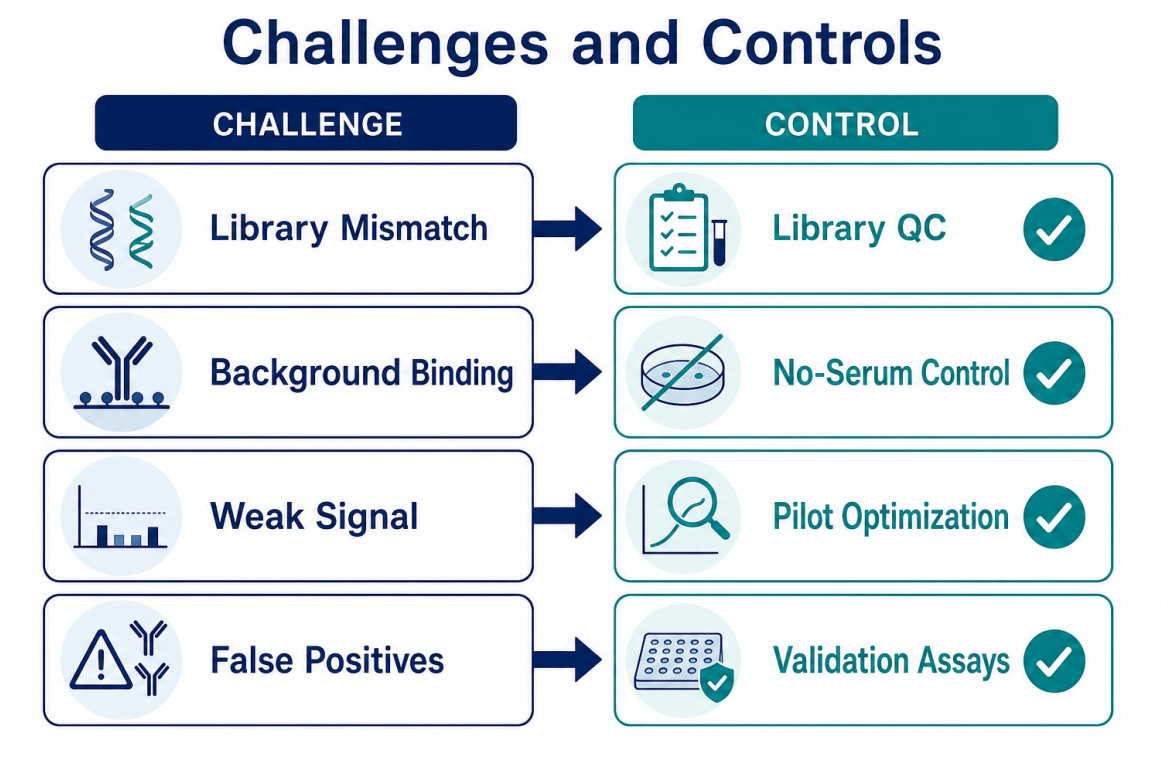
Figure 4. Technical challenges can be managed through library QC, matched controls, replicate checks, and validation planning.
How to Decide Whether the Assay Fits a Project
The assay is a strong fit when the research question is discovery-oriented, peptide-level antibody recognition is relevant, and the study can include proper controls. The method is especially useful when researchers need to screen many possible antigens, compare antibody profiles across groups, or identify candidate regions for downstream assays.
The screening method may not be the first choice when the antigen is already known and the goal is simple confirmation. A targeted assay may be more direct in that case. The method may also be limited when the key immune signal depends on conformational structure, glycosylation, lipid antigens, or native protein complexes.
When project teams are unsure whether the assay is the right fit, MtoZ Biolabs can review the sample type, library scope, cohort design, control plan, and validation strategy before full-scale screening begins.
Frequently Asked Questions
1. What does the assay measure?
The assay measures antibody binding to phage-displayed peptides. The method does not sequence antibody genes directly. The output is a peptide-level enrichment profile that reflects antibody reactivity against the represented library.
2. Is the method suitable for biomarker discovery?
Yes. The method can support serological biomarker discovery by comparing peptide enrichment between disease and control groups. Candidate biomarkers still require independent validation before diagnostic or translational use.
3. What sample types are commonly used?
Serum and plasma are the most common sample types because circulating antibodies are accessible. Other antibody-containing fluids may be possible if sample matrix, antibody abundance, and study goals are compatible.
4. Can the assay detect conformational epitopes?
Usually not directly. Phage-displayed peptide libraries are better suited for linear epitopes or motif-like recognition. Conformational epitopes usually require protein-based or structure-aware validation methods.
5. What makes a result reliable?
Reliable results depend on library fit, sample consistency, appropriate controls, replicate agreement, sequencing depth, background filtering, and validation planning. A single enriched peptide is not enough to establish a biological conclusion.
Conclusion
PhIP-Seq provides a scalable way to profile antibody reactivity across peptide libraries. The method supports epitope discovery, serological profiling, infectious disease research, autoimmune studies, vaccine response analysis, cancer immunology, and candidate biomarker discovery. The strongest studies connect method choice with a clear research question, well- matched samples, appropriate controls, interpretable data outputs, and validation assays.
For research teams evaluating PhIP-Seq methods, applications, and challenges, the next step is to match the peptide library and control plan to the biological question. Contact MtoZ Biolabs to discuss whether the assay is suitable for the sample set, expected antibody signal, and validation goals of the project.
How to order?

