





MSX-DIA Quantitative Proteomics Services
- Reduced co-fragmentation interference: Traditional DIA uses a fixed window, and multiple precursor ions co-fragment, resulting in increased data complexity; MSX-DIA reduces ion interference and improves the detection ability of low-abundance proteins through random window selection + optimized ion distribution.
- Improved low-abundance protein detection ability: In the plasma proteome, high-abundance proteins (such as albumin and globulin) will mask low-abundance protein signals, while MSX-DIA can accurately quantify low-abundance proteins in a wider dynamic range through intelligent window optimization and efficient deconvolution.
- Enhanced quantitative accuracy and data consistency: MSX-DIA combines artificial intelligence algorithms (AI-based Data Processing) to improve the confidence of quantitative proteins, making it suitable for large-scale clinical sample analysis.
MSX-DIA Quantitative Proteomics Services utilize the multiplexed data-independent acquisition (MSX-DIA) technique, developed by MacCoss et al., to enable high-throughput, high-sensitivity, and high-precision quantitative proteomic analysis. MSX-DIA is an optimized DIA mass spectrometry data acquisition strategy that introduces ion multiplexing technology to improve the throughput, sensitivity, and quantification accuracy of data-independent acquisition (DIA)-based proteomics. In standard DIA, multiple precursor ions are fragmented within fixed isolation windows, often leading to co-fragmentation, which can compromise quantification accuracy. By randomizing and optimizing the selection of precursor ions within the window, MSX-DIA reduces co-fragmentation interference in complex samples, improves the depth of peptide and protein identification, and enhances the accuracy of quantitative data. Leveraging a high-resolution mass spectrometry platform, MtoZ Biolabs provides MSX-DIA Quantitative Proteomics Services for large-scale proteomic quantification, low-abundance protein analysis, biomarker discovery, and precision medicine research.
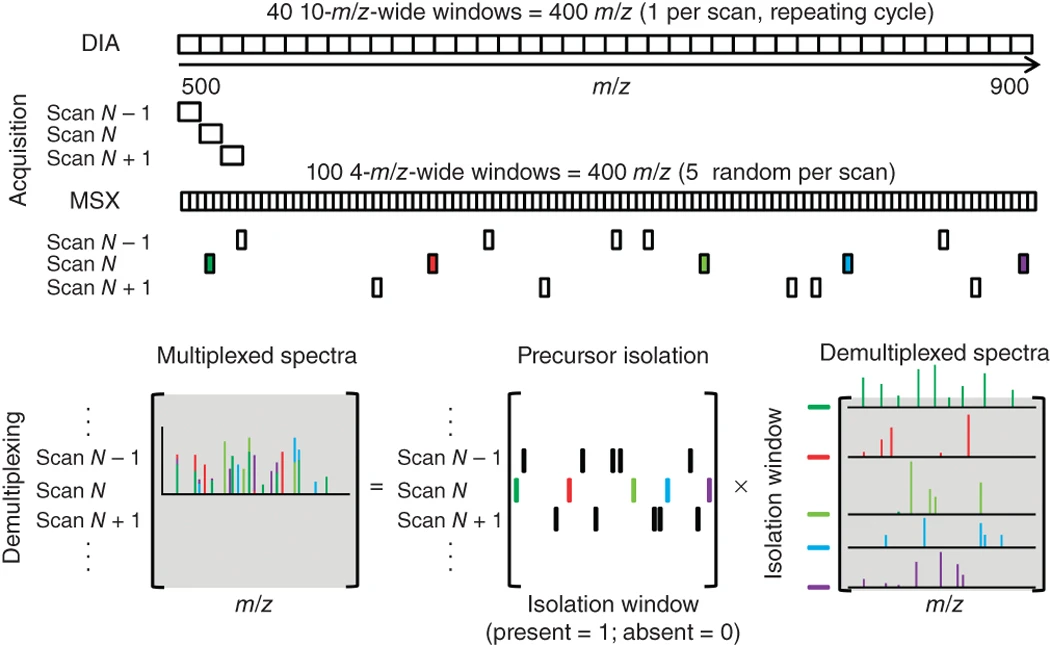
Egertson J D. et al. Nature Methods. 2013.
Analysis Workflow
1. Sample Preparation
Proteins are extracted from biological samples and digested using trypsin to generate high-quality peptides.
2. Liquid Chromatography-Mass Spectrometry (LC-MS/MS) Analysis
Peptides are separated using ultra-performance liquid chromatography (UPLC), and data acquisition is performed on a high-resolution mass spectrometer using the MSX-DIA acquisition mode.
3. Data Analysis (Deconvolution & Quantification)
Perform deconvolution, match databases, and accurately identify and quantify proteins.
4. Bioinformatics Analysis
Key proteins and their biological significance are explored through pathway enrichment analysis, protein interaction network analysis, and biomarker discovery.
Service Advantages
1. Advanced Analysis Platform: MtoZ Biolabs established an advanced MSX-DIA Quantitative Proteomics Services platform, guaranteeing reliable, fast, and highly accurate analysis service.
2. One-Time-Charge: Our pricing is transparent, no hidden fees or additional costs.
3. High-Data-Quality: Deep data coverage with strict data quality control. AI-powered bioinformatics platform integrates all MSX-DIA Quantitative Proteomics Services data, providing clients with a comprehensive data report.
4. Reduced Co-Fragmentation Interference: By leveraging multiplexing and deconvolution algorithms, MSX-DIA enhances data quality and quantification reliability.
Applications
MSX-DIA Quantitative Proteomics Services can be applied to various research fields, including but not limited to the following:
Large-Scale Protein Quantification
MSX-DIA optimizes ion selection, reduces co-fragmentation interference, and enhances data deconvolution, significantly improving the depth and accuracy of protein quantification.
Biomarker Discovery and Validation
By enabling large-scale quantitative proteomic analysis, MSX-DIA Quantitative Proteomics Services facilitate the identification and validation of disease-associated proteins, providing high-precision data support for biomarker discovery.
Mechanism of Action and Regulatory Studies
Combined with protein interaction and pathway analysis, MSX-DIA can reveal regulatory relationships between biomolecules, elucidates protein function mechanisms, and advances research in cell biology, developmental biology, and disease mechanisms.
Drug Target Research
MSX-DIA Quantitative Proteomics Services has high-throughput protein quantitative analysis capabilities, can identify disease-related target proteins, and analyze their dynamic changes, to assist in the screening of new drug targets.
FAQ
Q. Is MSX-DIA Superior to Standard DIA for Quantification in High-Complexity Samples (e.g., Plasma Proteomics)?
The quantitative ability of MSX-DIA in complex samples is significantly better than that of standard DIA, mainly reflected in:
In highly complex samples (such as plasma proteome), MSX-DIA has higher protein detection depth, stronger low-abundance protein quantification capability and more stable quantitative accuracy, and is one of the currently advanced DIA mass spectrometry quantitative strategies.
Q. Is MSX-DIA Suitable for Low-Abundance Protein Quantification? How Can Its Detection Sensitivity Be Improved?
MSX-DIA outperforms standard DIA in low-abundance protein quantification and its detection sensitivity can be enhanced through the following strategies:
Optimized Sample Preparation:Depleting high-abundance proteins (e.g., albumin in plasma) increases the signal intensity of low-abundance proteins.
Enhanced LC-MS Resolution:Utilizing ultra-performance liquid chromatography (UPLC) and high-resolution mass spectrometry (e.g., Orbitrap Eclipse) improves peptide separation efficiency and enhances signal intensity.
Optimized Data Processing Workflow:Implementing DIA-NN or Spectronaut, combined with a high-quality spectral library, reduces false positives and ensures reliable quantification of low-abundance proteins.
Increased Sampling Depth (Deep Scanning):Employing multiple DIA scans (deep scanning) enhances low-abundance protein signal coverage, improving detection depth.
Deliverables
1. Comprehensive Experimental Details
2. Materials, Instruments, and Methods
3. Total Ion Chromatogram & Quality Control Assessment (project-dependent)
4. Data Analysis, Preprocessing, and Estimation (project-dependent)
5. Bioinformatics Analysis
6. Raw Data Files
Case Study
This study utilized MSX-DIA Quantitative Proteomics technology to analyze the proteomics of psychotic experiences (PEs) focusing on the complement pathway. The findings revealed that changes in complement system protein expression at age 12 were significantly associated with the risk of psychotic experiences at age 18. This suggested that the complement system may play a crucial role in the early progression of psychiatric disorders.
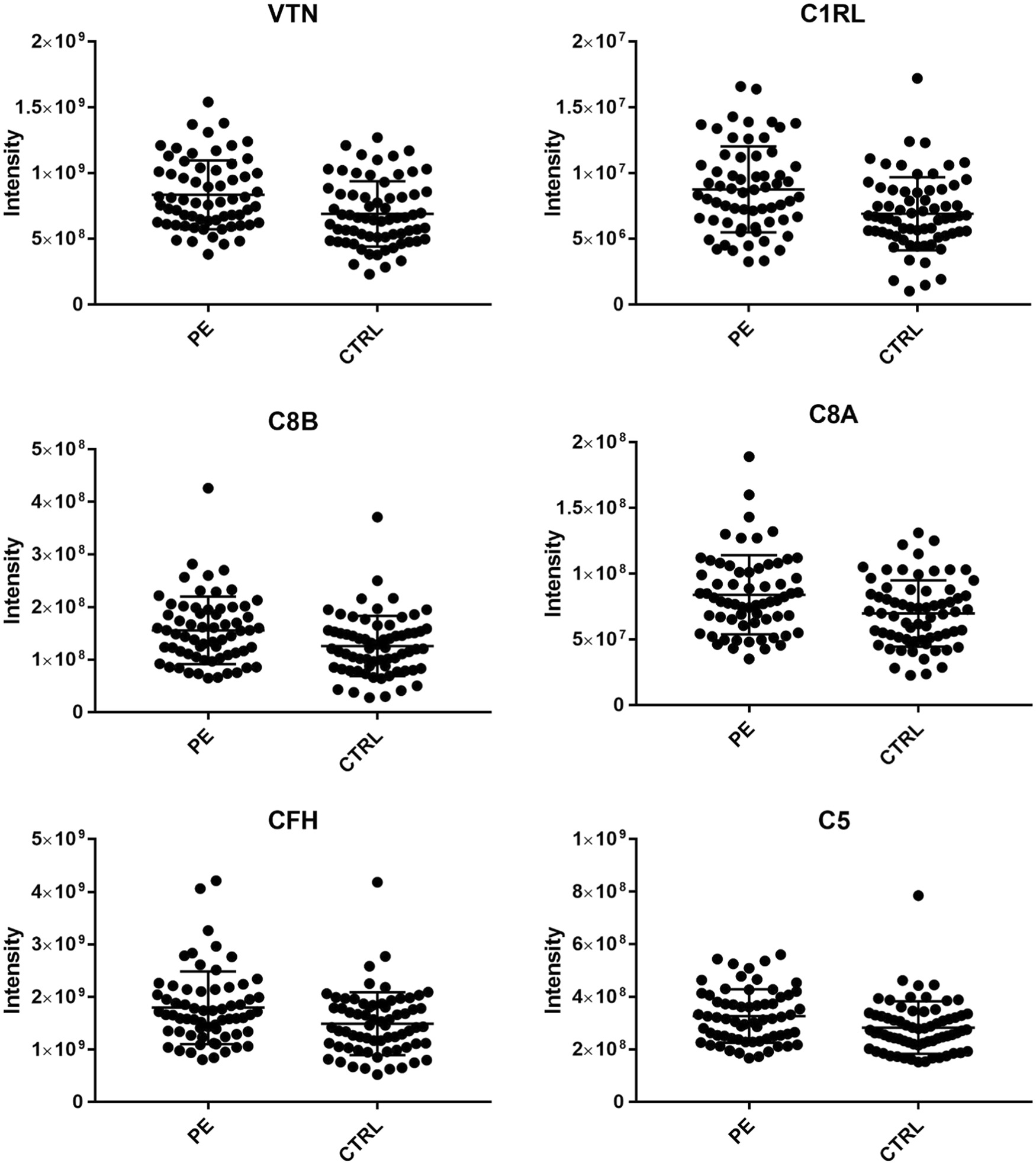
Föcking M. et al. Mol Psychiatry. 2021.
How to order?

